Department of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, North Carolina, United States of America.
Department of Cell Biology, Duke University School of Medicine, Durham, North Carolina, United States of America.
PLoS Comput Biol. 2022 Sep 12;18(9):e1009921. doi: 10.1371/journal.pcbi.1009921. eCollection 2022 Sep.
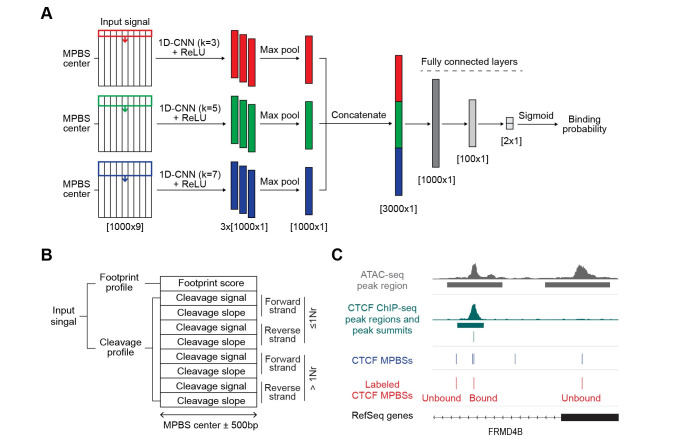
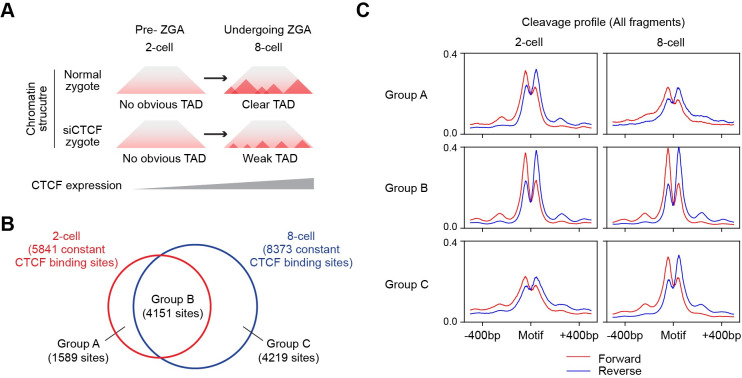
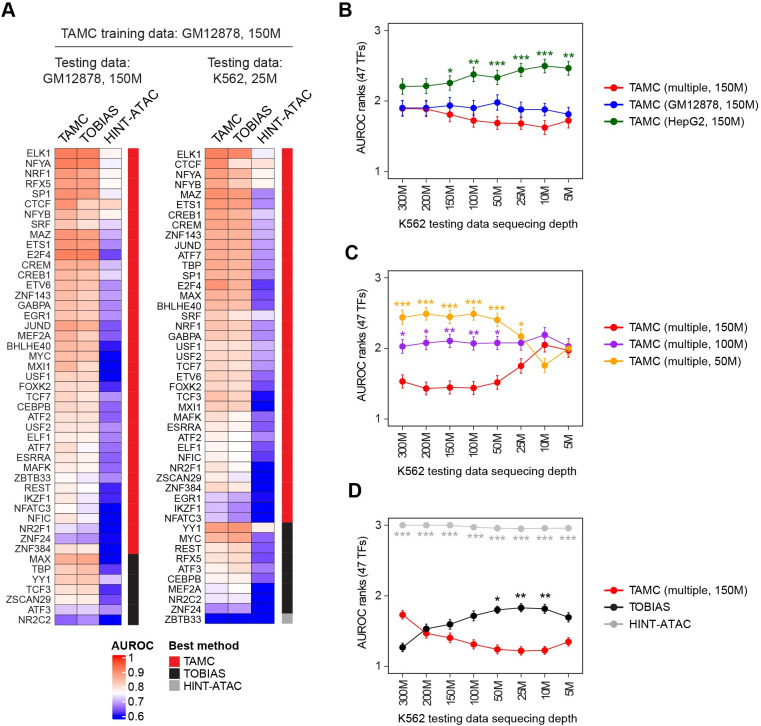
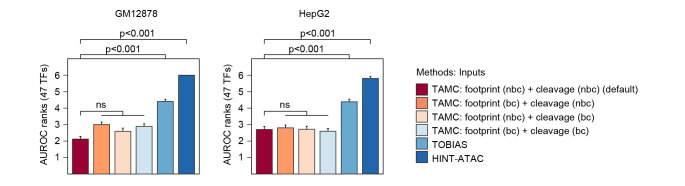
Determining transcriptional factor binding sites (TFBSs) is critical for understanding the molecular mechanisms regulating gene expression in different biological conditions. Biological assays designed to directly mapping TFBSs require large sample size and intensive resources. As an alternative, ATAC-seq assay is simple to conduct and provides genomic cleavage profiles that contain rich information for imputing TFBSs indirectly. Previous footprint-based tools are inheritably limited by the accuracy of their bias correction algorithms and the efficiency of their feature extraction models. Here we introduce TAMC (Transcriptional factor binding prediction from ATAC-seq profile at Motif-predicted binding sites using Convolutional neural networks), a deep-learning approach for predicting motif-centric TF binding activity from paired-end ATAC-seq data. TAMC does not require bias correction during signal processing. By leveraging a one-dimensional convolutional neural network (1D-CNN) model, TAMC make predictions based on both footprint and non-footprint features at binding sites for each TF and outperforms existing footprinting tools in TFBS prediction particularly for ATAC-seq data with limited sequencing depth.
确定转录因子结合位点(TFBSs)对于理解不同生物条件下基因表达的分子机制至关重要。旨在直接绘制 TFBSs 的生物学检测需要大量样本和密集资源。作为替代方案,ATAC-seq 检测方法简单易行,并提供了丰富的基因组切割谱,可间接提供丰富的 TFBS 推断信息。以前基于足迹的工具固有地受到其偏差校正算法的准确性和特征提取模型的效率的限制。在这里,我们介绍了 TAMC(使用卷积神经网络从 motif-predicted 结合位点的 ATAC-seq 图谱中预测转录因子结合预测),这是一种从配对末端 ATAC-seq 数据中预测以基序为中心的 TF 结合活性的深度学习方法。TAMC 在信号处理过程中不需要偏差校正。通过利用一维卷积神经网络(1D-CNN)模型,TAMC 可以基于每个 TF 的结合位点的足迹和非足迹特征进行预测,并且在 TFBS 预测方面优于现有的足迹检测工具,特别是对于测序深度有限的 ATAC-seq 数据。