Department of Medicine-Division of Medical Oncology, University of Colorado-Anschutz Medical Campus, Aurora, Colorado, USA.
Department of Pathology, University of Colorado-Anschutz Medical Campus, Aurora, Colorado, USA.
Thorac Cancer. 2022 Nov;13(21):3032-3041. doi: 10.1111/1759-7714.14656. Epub 2022 Sep 13.
ROS1 tyrosine kinase inhibitors (TKIs) have demonstrated significant clinical benefit for ROS1+ NSCLC patients. However, TKI resistance inevitably develops through ROS1 kinase domain (KD) modification or another kinase driving bypass signaling. While multiple TKIs have been designed to target ROS1 KD mutations, less is known about bypass signaling in TKI-resistant ROS1+ lung cancers.
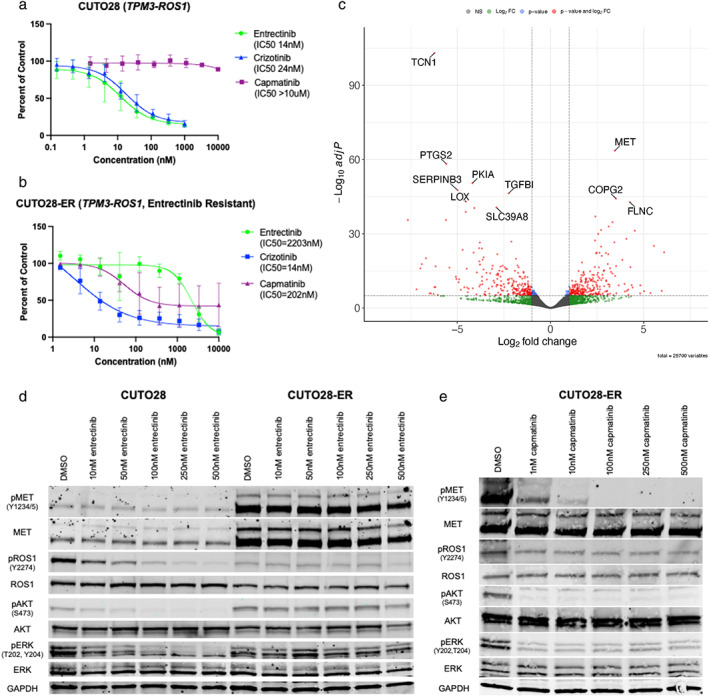
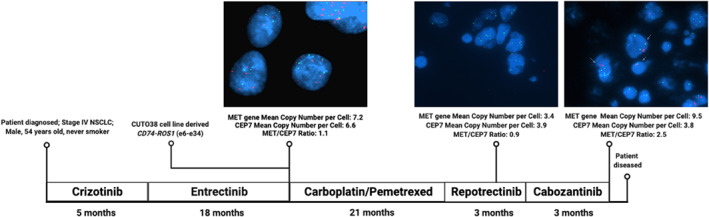
Utilizing a primary, patient-derived TPM3-ROS1 cell line (CUTO28), we derived an entrectinib-resistant line (CUTO28-ER). We evaluated proliferation and signaling responses to TKIs, and utilized RNA sequencing, whole exome sequencing, and fluorescence in situ hybridization to detect transcriptional, mutational, and copy number alterations, respectively. We substantiated in vitro findings using a CD74-ROS1 NSCLC patient's tumor samples. Last, we analyzed circulating tumor DNA (ctDNA) from ROS1+ NSCLC patients in the STARTRK-2 entrectinib trial to determine the prevalence of MET amplification.
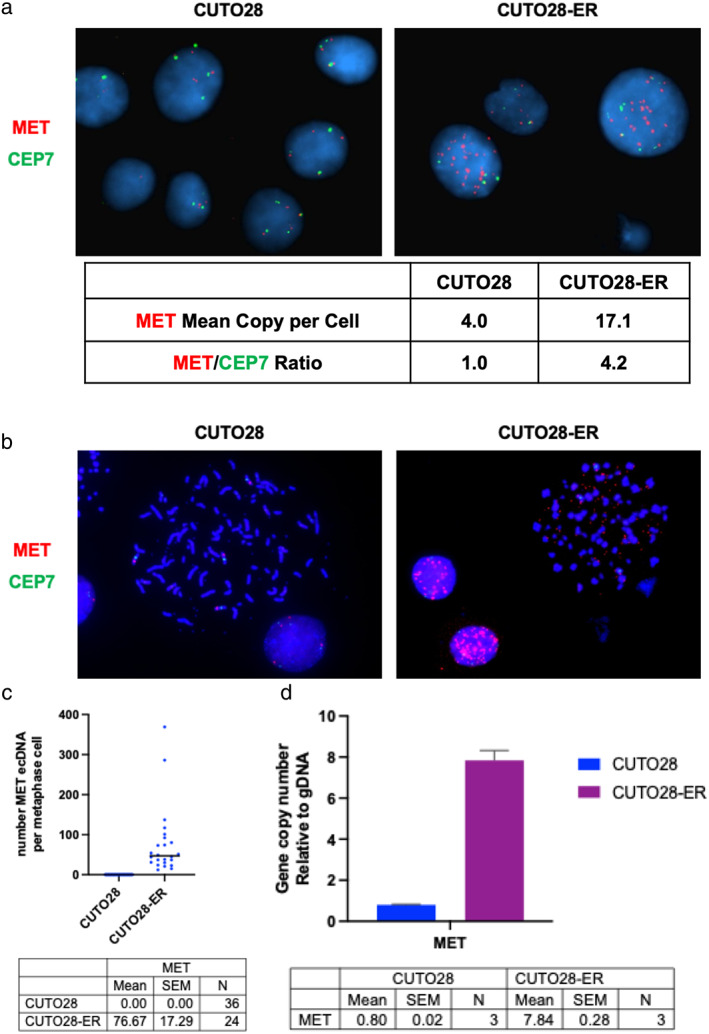
CUTO28-ER cells did not exhibit ROS1 KD mutations. MET TKIs inhibited proliferation and downstream signaling and MET transcription was elevated in CUTO28-ER cells. CUTO28-ER cells displayed extrachromosomal (ecDNA) MET amplification without MET activating mutations, exon 14 skipping, or fusions. The CD74-ROS1 patient samples illustrated MET amplification while receiving ROS1 TKI. Finally, two of 105 (1.9%) entrectinib-resistant ROS1+ NSCLC STARTRK-2 patients with ctDNA analysis at enrollment and disease progression displayed MET amplification.
Treatment with ROS1-selective inhibitors may lead to MET-mediated resistance. The discovery of ecDNA MET amplification is noteworthy, as ecDNA is associated with more aggressive cancers. Following progression on ROS1-selective inhibitors, MET gene testing and treatments targeting MET should be explored to overcome MET-driven resistance.
ROS1 酪氨酸激酶抑制剂(TKI)为 ROS1+ NSCLC 患者带来了显著的临床获益。然而,ROS1 激酶结构域(KD)的改变或其他激酶驱动旁路信号转导,不可避免地会导致 TKI 耐药。虽然已经设计了多种 TKI 来靶向 ROS1 KD 突变,但对于 TKI 耐药的 ROS1+ 肺癌中的旁路信号转导知之甚少。
我们利用原发性、患者源性的 TPM3-ROS1 细胞系(CUTO28),衍生出恩曲替尼耐药株(CUTO28-ER)。我们评估了 TKI 对增殖和信号转导的影响,并分别利用 RNA 测序、全外显子组测序和荧光原位杂交来检测转录、突变和拷贝数改变。我们使用 CD74-ROS1 NSCLC 患者的肿瘤样本验证了体外研究结果。最后,我们分析了 STARTRK-2 恩曲替尼试验中 ROS1+ NSCLC 患者的循环肿瘤 DNA(ctDNA),以确定 MET 扩增的发生率。
CUTO28-ER 细胞未出现 ROS1 KD 突变。MET TKI 抑制了增殖和下游信号转导,并且 CUTO28-ER 细胞中的 MET 转录上调。CUTO28-ER 细胞显示了额外染色体(ecDNA)MET 扩增,而没有 MET 激活突变、外显子 14 跳跃或融合。CD74-ROS1 患者样本在接受 ROS1 TKI 治疗时显示出 MET 扩增。最后,在纳入和疾病进展时进行 ctDNA 分析的 105 名恩曲替尼耐药 ROS1+ NSCLC STARTRK-2 患者中,有 2 名(1.9%)出现 MET 扩增。
ROS1 选择性抑制剂的治疗可能导致 MET 介导的耐药。ecDNA MET 扩增的发现值得注意,因为 ecDNA 与更具侵袭性的癌症有关。在 ROS1 选择性抑制剂进展后,应探索 MET 基因检测和针对 MET 的治疗方法,以克服 MET 驱动的耐药。