Department of Biology, University of Puerto Rico-Rio Piedras, San Juan PR 00931, Puerto Rico.
Department of Biological Sciences, The George Washington University, Washington, DC 20052, USA.
Genome Res. 2022 Oct;32(10):1862-1875. doi: 10.1101/gr.276839.122. Epub 2022 Sep 15.
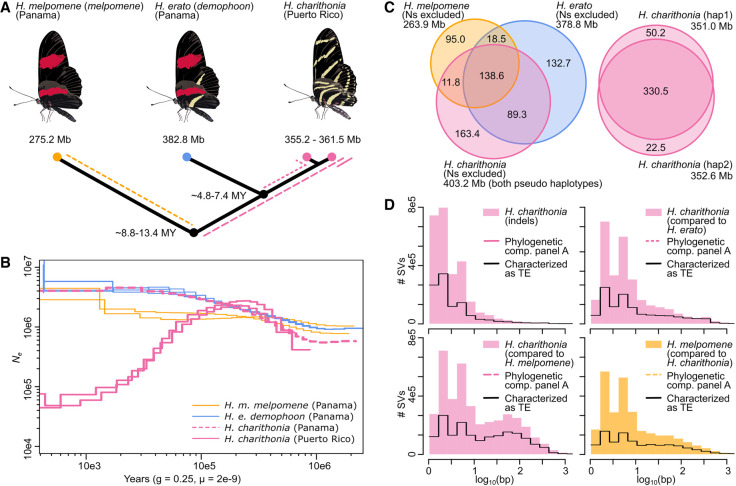
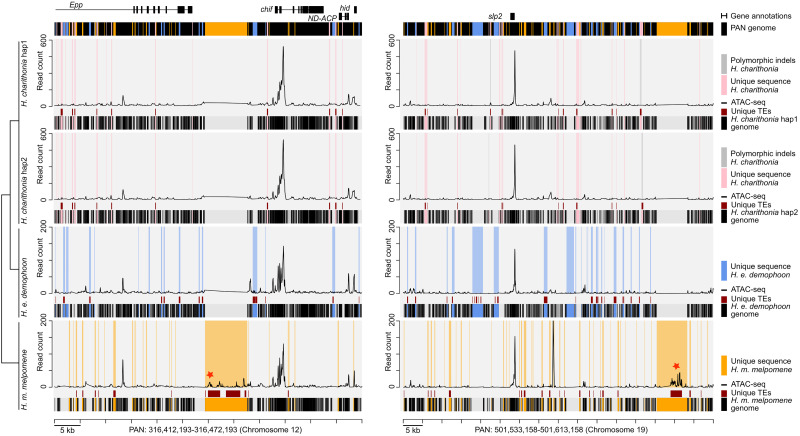
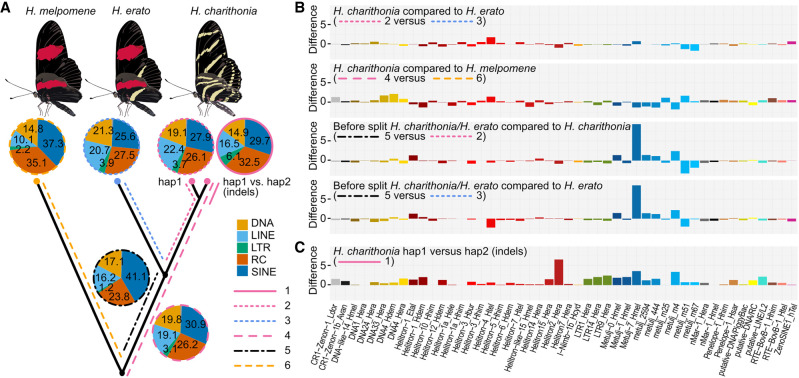
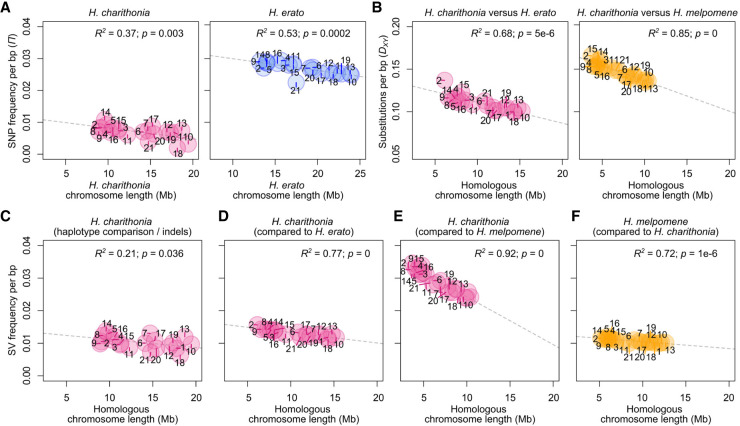
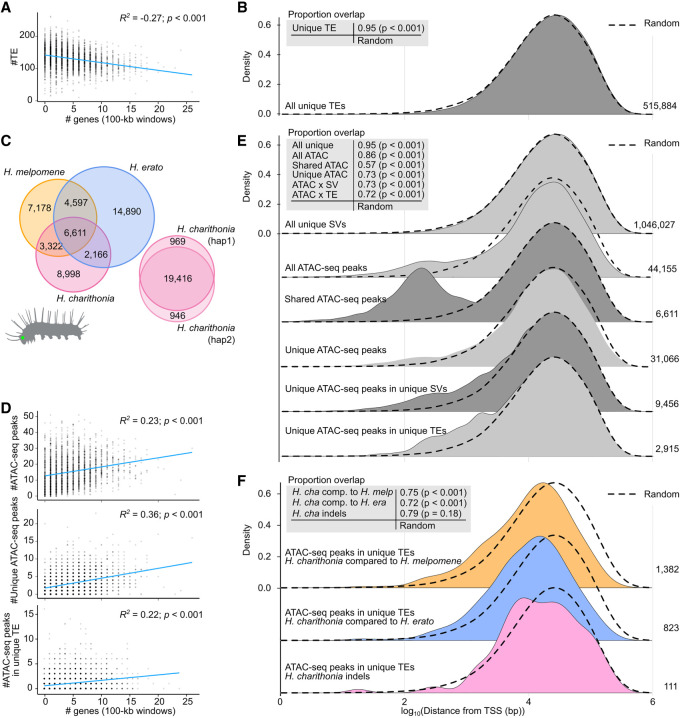
Despite insertions and deletions being the most common structural variants (SVs) found across genomes, not much is known about how much these SVs vary within populations and between closely related species, nor their significance in evolution. To address these questions, we characterized the evolution of indel SVs using genome assemblies of three closely related butterfly species. Over the relatively short evolutionary timescales investigated, up to 18.0% of the genome was composed of indels between two haplotypes of an individual butterfly and up to 62.7% included lineage-specific SVs between the genomes of the most distant species (11 Mya). Lineage-specific sequences were mostly characterized as transposable elements (TEs) inserted at random throughout the genome and their overall distribution was similarly affected by linked selection as single nucleotide substitutions. Using chromatin accessibility profiles (i.e., ATAC-seq) of head tissue in caterpillars to identify sequences with potential -regulatory function, we found that out of the 31,066 identified differences in chromatin accessibility between species, 30.4% were within lineage-specific SVs and 9.4% were characterized as TE insertions. These TE insertions were localized closer to gene transcription start sites than expected at random and were enriched for sites with significant resemblance to several transcription factor binding sites with known function in neuron development in We also identified 24 TE insertions with head-specific chromatin accessibility. Our results show high rates of structural genome evolution that were previously overlooked in comparative genomic studies and suggest a high potential for structural variation to serve as raw material for adaptive evolution.
尽管插入和缺失是在基因组中发现的最常见的结构变体 (SV),但对于这些 SV 在种群内和密切相关物种之间的变化程度及其在进化中的意义知之甚少。为了解决这些问题,我们使用三种密切相关的蝴蝶物种的基因组组装来描述插入缺失 SV 的进化。在所研究的相对较短的进化时间尺度内,多达 18.0%的基因组由个体蝴蝶的两个单倍型之间的插入缺失组成,多达 62.7%的基因组包括最遥远物种之间的谱系特异性 SV (11 Mya)。谱系特异性序列主要特征为随机插入基因组中的转座元件 (TEs),它们的整体分布同样受到连锁选择的影响,就像单核苷酸替换一样。使用毛毛虫头部组织的染色质可及性图谱 (即 ATAC-seq) 来识别具有潜在 -调控功能的序列,我们发现,在物种间的 31066 个鉴定出的染色质可及性差异中,30.4%存在于谱系特异性 SV 中,9.4%被表征为 TE 插入。这些 TE 插入比随机预期更靠近基因转录起始位点,并富集了与几个转录因子结合位点具有显著相似性的位点,这些转录因子结合位点在神经元发育中具有已知功能。我们还鉴定了 24 个具有头部特异性染色质可及性的 TE 插入。我们的研究结果表明,结构基因组进化的速度很高,这在以前的比较基因组研究中被忽视了,这表明结构变异有很高的潜力成为适应进化的原始材料。