Department of Pediatric Cardiology, Xinhua Hospital, Affiliated to Shanghai Jiao Tong University School of Medicine, Room 505, Scientific Building, Shanghai, 200092, China.
Department of Pediatrics, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, 450052, China.
Hum Genomics. 2022 Sep 19;16(1):41. doi: 10.1186/s40246-022-00409-9.
Heterotaxy syndrome (HTX) is caused by aberrant left-right patterning early in embryonic development, which results in abnormal positioning and morphology of the thoracic and abdominal organs. Currently, genetic testing discerns the underlying genetic cause in less than 20% of sporadic HTX cases, indicating that genetic pathogenesis remains poorly understood. In this study, we aim to garner a deeper understanding of the genetic factors of this disease by documenting the effect of different matrix metalloproteinase 21 (MMP21) variants on disease occurrence and pathogenesis.
Eighty-one HTX patients with complex congenital heart defects and 89 healthy children were enrolled, and we investigated the pathogenetic variants related to patients with HTX by exome sequencing. Zebrafish splice-blocking Morpholino oligo-mediated transient suppression assays were performed to confirm the potential pathogenicity of missense variants found in these patients with HTX.
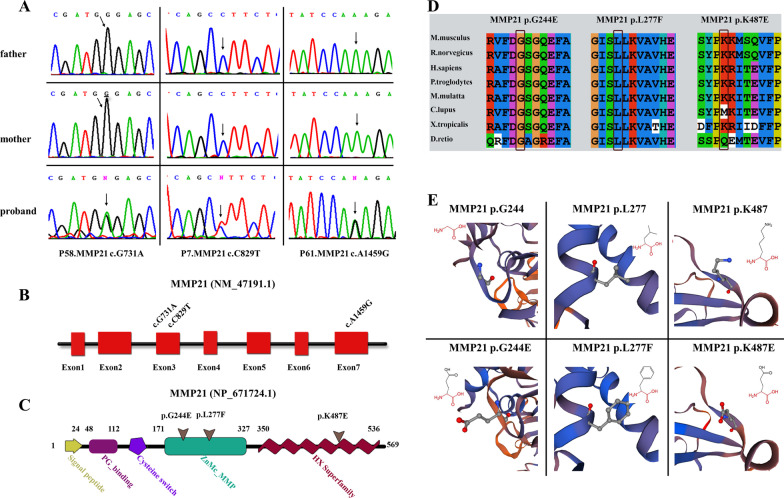
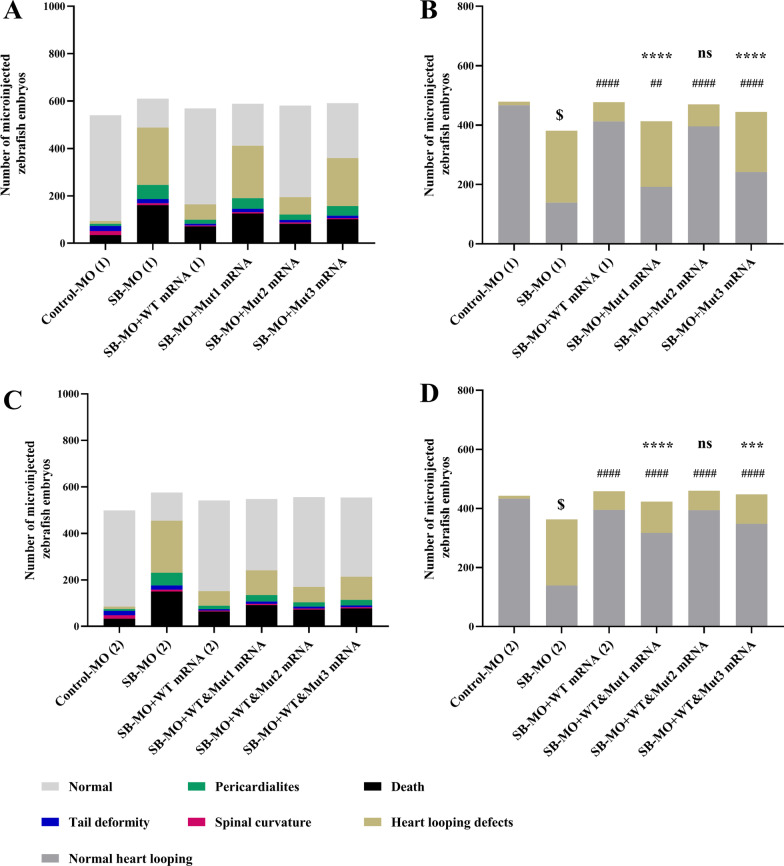
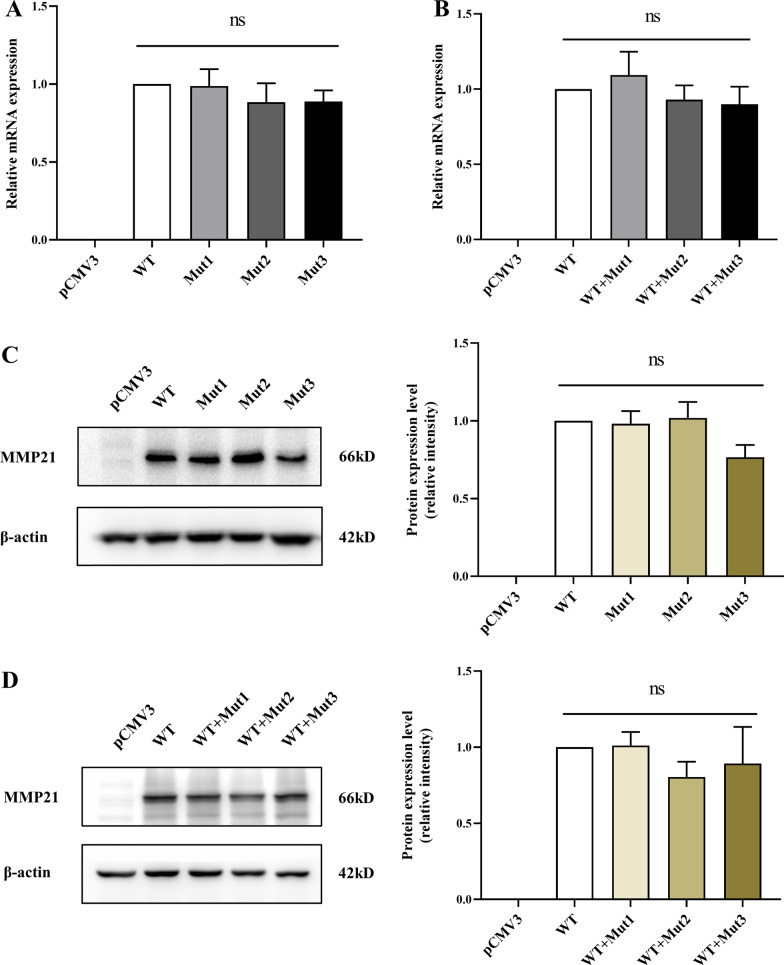
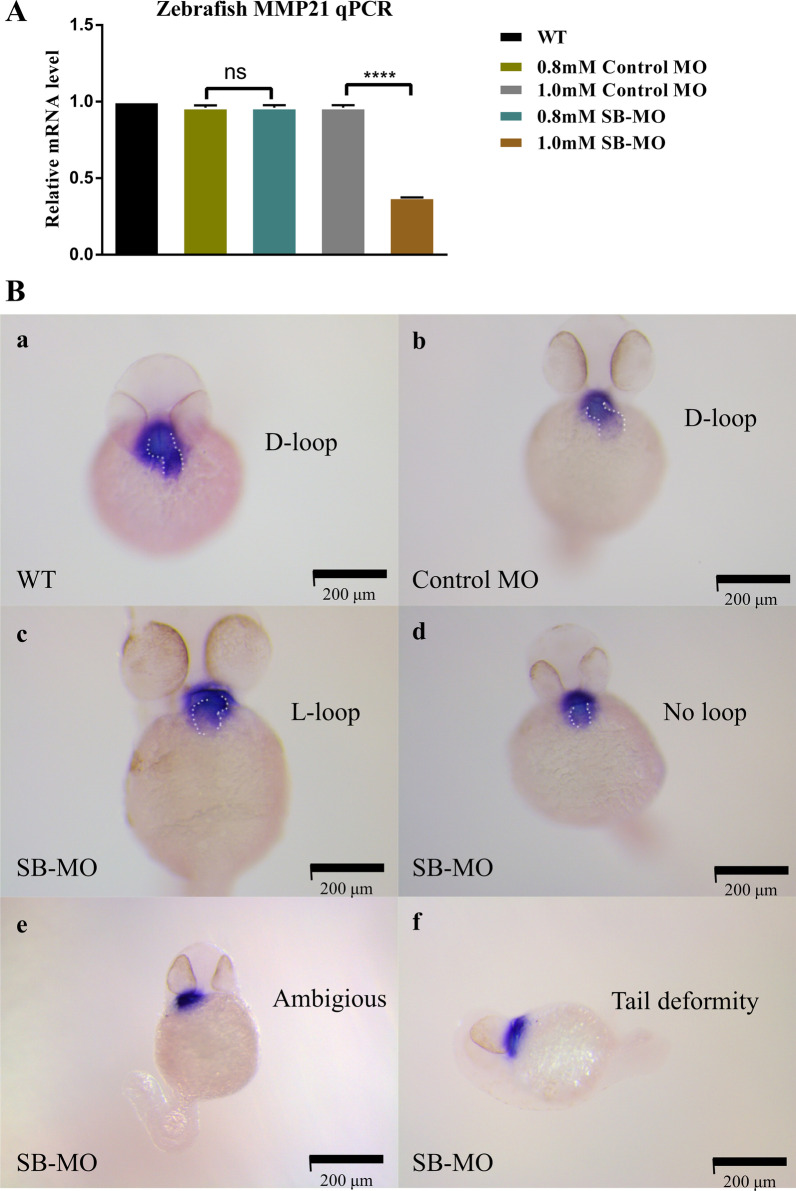
Three MMP21 heterozygous non-synonymous variants (c.731G > A (p.G244E), c.829C > T (p.L277F), and c.1459A > G (p.K487E)) were identified in three unrelated Chinese Han patients with HTX and complex congenital heart defects. Sanger sequencing confirmed that all variants were de novo. Cell transfection assay showed that none of the variants affect mRNA and protein expression levels of MMP21. Knockdown expression of mmp21 by splice-blocking Morpholino oligo in zebrafish embryos revealed a heart looping disorder, and mutant human MMP21 mRNA (c.731G > A, c.1459A > G, heterozygous mRNA (wild-type&c.731G > A), as well as heterozygous mRNA (wild-type& c.1459A > G) could not effectively rescue the heart looping defects. A patient with the MMP21 p.G244E variant was identified with other potential HTX-causing missense mutations, whereas the patient with the MMP21 p.K487E variant had no genetic mutations in other causative genes related to HTX.
Our study highlights the role of the disruptive heterozygous MMP21 variant (p.K487E) in the etiology of HTX with complex cardiac malformations and expands the current mutation spectrum of MMP21 in HTX.
异构综合征(HTX)是由胚胎发育早期左右模式的异常引起的,导致胸腹部器官的位置和形态异常。目前,遗传检测在不到 20%的散发性 HTX 病例中辨别出潜在的遗传原因,表明遗传发病机制仍知之甚少。在这项研究中,我们旨在通过记录不同基质金属蛋白酶 21(MMP21)变体对疾病发生和发病机制的影响,更深入地了解该疾病的遗传因素。
纳入 81 例伴复杂先天性心脏病的 HTX 患者和 89 例健康儿童,通过外显子组测序研究与 HTX 患者相关的致病变体。通过斑马鱼剪接阻断 Morpholino 寡核苷酸介导的瞬时抑制试验,证实了在这些 HTX 患者中发现的错义变体的潜在致病性。
在 3 例无亲缘关系的中国汉族 HTX 伴复杂先天性心脏病患者中发现了 3 种 MMP21 杂合性非同义变异(c.731G > A(p.G244E),c.829C > T(p.L277F)和 c.1459A > G(p.K487E))。Sanger 测序证实所有变异均为新生。细胞转染试验表明,这些变异均不影响 MMP21 的 mRNA 和蛋白表达水平。在斑马鱼胚胎中,剪接阻断 Morpholino 寡核苷酸敲低 mmp21 的表达,导致心脏环化障碍,而突变型人 MMP21 mRNA(c.731G > A、c.1459A > G、杂合性 mRNA(野生型&c.731G > A)和杂合性 mRNA(野生型&c.1459A > G)均不能有效挽救心脏环化缺陷。一位 MMP21 p.G244E 变异患者还发现了其他潜在的 HTX 致病错义突变,而 MMP21 p.K487E 变异患者在其他与 HTX 相关的致病基因中没有遗传突变。
本研究强调了破坏性杂合 MMP21 变异(p.K487E)在伴有复杂心脏畸形的 HTX 发病机制中的作用,并扩展了 MMP21 在 HTX 中的当前突变谱。