Thellung Stefano, Corsaro Alessandro, Dellacasagrande Irene, Nizzari Mario, Zambito Martina, Florio Tullio
Section of Pharmacology, Department of Internal Medicine (DiMI), University of Genova, Genova, Italy.
IRCCS Ospedale Policlinico San Martino, Genova, Italy.
Front Neurosci. 2022 Sep 6;16:966019. doi: 10.3389/fnins.2022.966019. eCollection 2022.
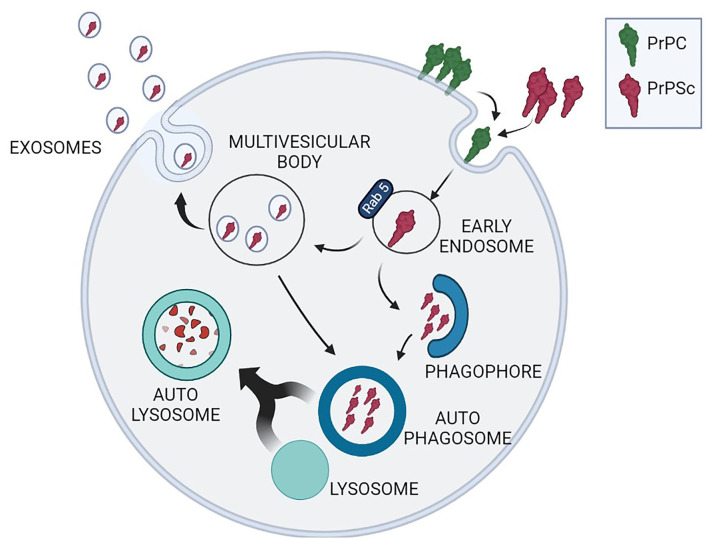
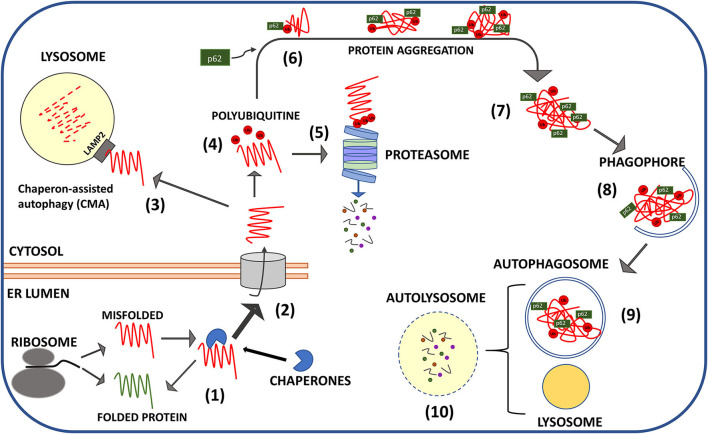
Transmissible spongiform encephalopathies (TSEs), or prion diseases, are progressive neurodegenerative disorders of the central nervous system that affect humans and animals as sporadic, inherited, and infectious forms. Similarly to Alzheimer's disease and other neurodegenerative disorders, any attempt to reduce TSEs' lethality or increase the life expectancy of affected individuals has been unsuccessful. Typically, the onset of symptoms anticipates the fatal outcome of less than 1 year, although it is believed to be the consequence of a decades-long process of neuronal death. The duration of the symptoms-free period represents by itself a major obstacle to carry out effective neuroprotective therapies. Prions, the infectious entities of TSEs, are composed of a protease-resistant protein named prion protein scrapie (PrP) from the prototypical TSE form that afflicts ovines. PrP misfolding from its physiological counterpart, cellular prion protein (PrP), is the unifying pathogenic trait of all TSEs. PrP is resistant to intracellular turnover and undergoes amyloid-like fibrillation passing through the formation of soluble dimers and oligomers, which are likely the effective neurotoxic entities. The failure of PrP removal is a key pathogenic event that defines TSEs as proteopathies, likewise other neurodegenerative disorders, including Alzheimer's, Parkinson's, and Huntington's disease, characterized by alteration of proteostasis. Under physiological conditions, protein quality control, led by the ubiquitin-proteasome system, and macroautophagy clears cytoplasm from improperly folded, redundant, or aggregation-prone proteins. There is evidence that both of these crucial homeostatic pathways are impaired during the development of TSEs, although it is still unclear whether proteostasis alteration facilitates prion protein misfolding or, rather, PrP protease resistance hampers cytoplasmic protein quality control. This review is aimed to critically analyze the most recent advancements in the cause-effect correlation between PrP misfolding and proteostasis alterations and to discuss the possibility that pharmacological restoring of ubiquitin-proteasomal competence and stimulation of autophagy could reduce the intracellular burden of PrP and ameliorate the severity of prion-associated neurodegeneration.
传染性海绵状脑病(TSEs),即朊病毒病,是中枢神经系统的进行性神经退行性疾病,以散发性、遗传性和传染性形式影响人类和动物。与阿尔茨海默病和其他神经退行性疾病类似,任何降低TSEs致死率或延长受影响个体预期寿命的尝试都未成功。通常,症状出现后不到1年就会导致死亡,尽管这被认为是长达数十年的神经元死亡过程的结果。无症状期的持续时间本身就是实施有效神经保护疗法的主要障碍。朊病毒是TSEs的感染性实体,由一种名为羊瘙痒病朊病毒蛋白(PrP)的抗蛋白酶蛋白组成,该蛋白来自影响绵羊的典型TSE形式。PrP从其生理对应物细胞朊病毒蛋白(PrPC)错误折叠是所有TSEs的统一致病特征。PrP对细胞内周转具有抗性,并通过形成可溶性二聚体和寡聚体经历淀粉样纤维化,这些可能是有效的神经毒性实体。PrP清除失败是一个关键的致病事件,将TSEs定义为蛋白病,同样还有其他神经退行性疾病,包括阿尔茨海默病、帕金森病和亨廷顿病,其特征是蛋白质稳态改变。在生理条件下,由泛素-蛋白酶体系统主导的蛋白质质量控制和巨自噬清除细胞质中错误折叠、多余或易于聚集的蛋白质。有证据表明,在TSEs发展过程中,这两个关键的稳态途径均受损,尽管目前尚不清楚蛋白质稳态改变是促进朊病毒蛋白错误折叠,还是PrP蛋白酶抗性阻碍细胞质蛋白质质量控制。本综述旨在批判性地分析PrP错误折叠与蛋白质稳态改变之间因果关系的最新进展,并讨论通过药理学方法恢复泛素-蛋白酶体功能和刺激自噬是否有可能减轻细胞内PrP负担并改善朊病毒相关神经退行性变的严重程度。