Transversal activities in Applied Genomics, Sciensano, Juliette Wytsmanstraat 14, Brussels, Belgium.
National Influenza Centre, Sciensano, Juliette Wytsmanstraat 14, Brussels, Belgium.
Microb Genom. 2022 Sep;8(9). doi: 10.1099/mgen.0.000867.
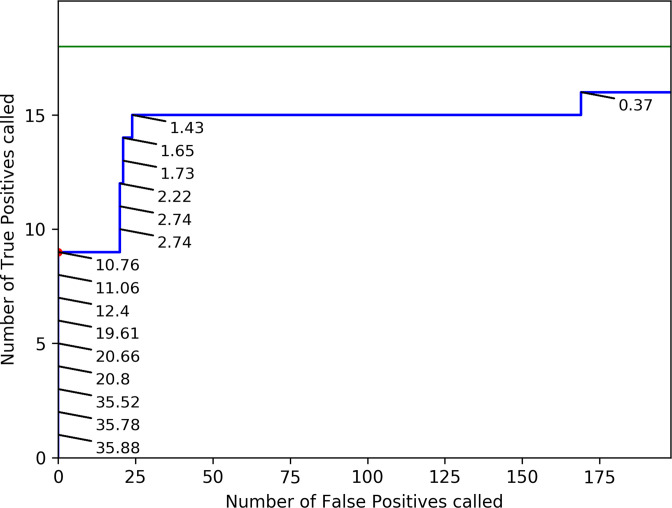
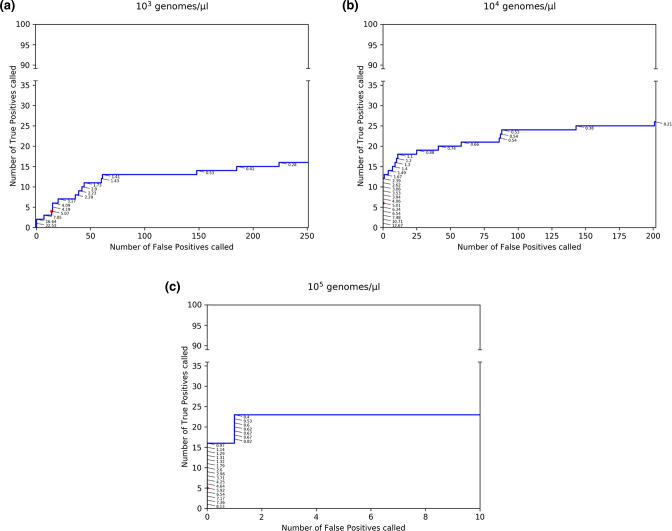
Influenza viruses exhibit considerable diversity between hosts. Additionally, different quasispecies can be found within the same host. High-throughput sequencing technologies can be used to sequence a patient-derived virus population at sufficient depths to identify low-frequency variants (LFV) present in a quasispecies, but many challenges remain for reliable LFV detection because of experimental errors introduced during sample preparation and sequencing. High genomic copy numbers and extensive sequencing depths are required to differentiate false positive from real LFV, especially at low allelic frequencies (AFs). This study proposes a general approach for identifying LFV in patient-derived samples obtained during routine surveillance. Firstly, validated thresholds were determined for LFV detection, whilst balancing both the cost and feasibility of reliable LFV detection in clinical samples. Using a genetically well-defined population of influenza A viruses, thresholds of at least 10 genomes per microlitre and AF of ≥5 % were established as detection limits. Secondly, a subset of 59 retained influenza A (H3N2) samples from the 2016-2017 Belgian influenza season was composed. Thirdly, as a proof of concept for the added value of LFV for routine influenza monitoring, potential associations between patient data and whole genome sequencing data were investigated. A significant association was found between a high prevalence of LFV and disease severity. This study provides a general methodology for influenza LFV detection, which can also be adopted by other national influenza reference centres and for other viruses such as SARS-CoV-2. Additionally, this study suggests that the current relevance of LFV for routine influenza surveillance programmes might be undervalued.
流感病毒在宿主间表现出相当大的多样性。此外,同一宿主内也可以发现不同的准种。高通量测序技术可用于对患者来源的病毒群体进行足够深度的测序,以鉴定准种中存在的低频变异体(LFV),但由于在样品制备和测序过程中引入了实验误差,可靠的 LFV 检测仍存在许多挑战。需要高基因组拷贝数和广泛的测序深度来区分假阳性和真正的 LFV,尤其是在低等位基因频率(AF)时。本研究提出了一种用于识别常规监测中患者来源样本中 LFV 的通用方法。首先,确定了 LFV 检测的验证阈值,同时平衡了在临床样本中可靠检测 LFV 的成本和可行性。使用遗传上定义明确的流感 A 病毒群体,确定了至少 10 个基因组/微升和 AF≥5%的阈值作为检测限。其次,由 2016-2017 年比利时流感季节的 59 个保留的流感 A(H3N2)样本组成一个子集。第三,作为 LFV 对常规流感监测的附加价值的概念验证,研究了患者数据和全基因组测序数据之间的潜在关联。发现 LFV 高流行率与疾病严重程度之间存在显著关联。本研究提供了一种用于流感 LFV 检测的通用方法,其他国家流感参考中心和其他病毒(如 SARS-CoV-2)也可以采用该方法。此外,本研究表明,当前 LFV 对常规流感监测计划的相关性可能被低估。