National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA.
Bioscience Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA.
Viruses. 2024 Mar 11;16(3):430. doi: 10.3390/v16030430.
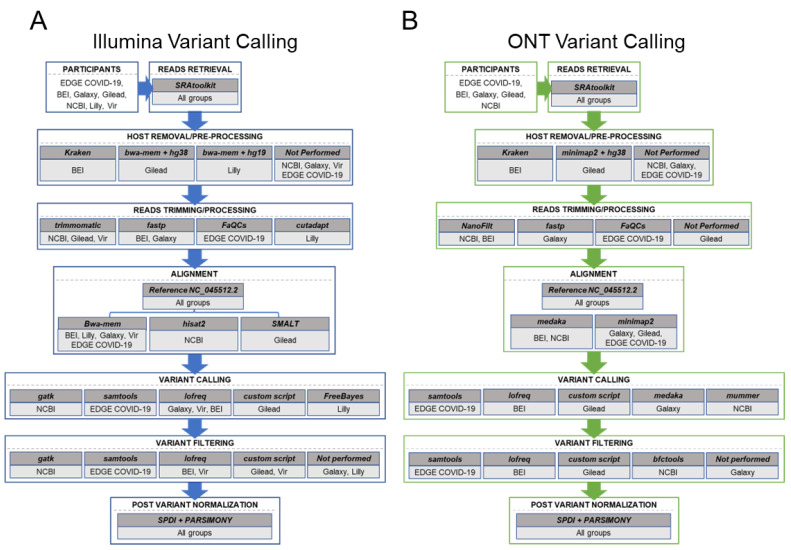
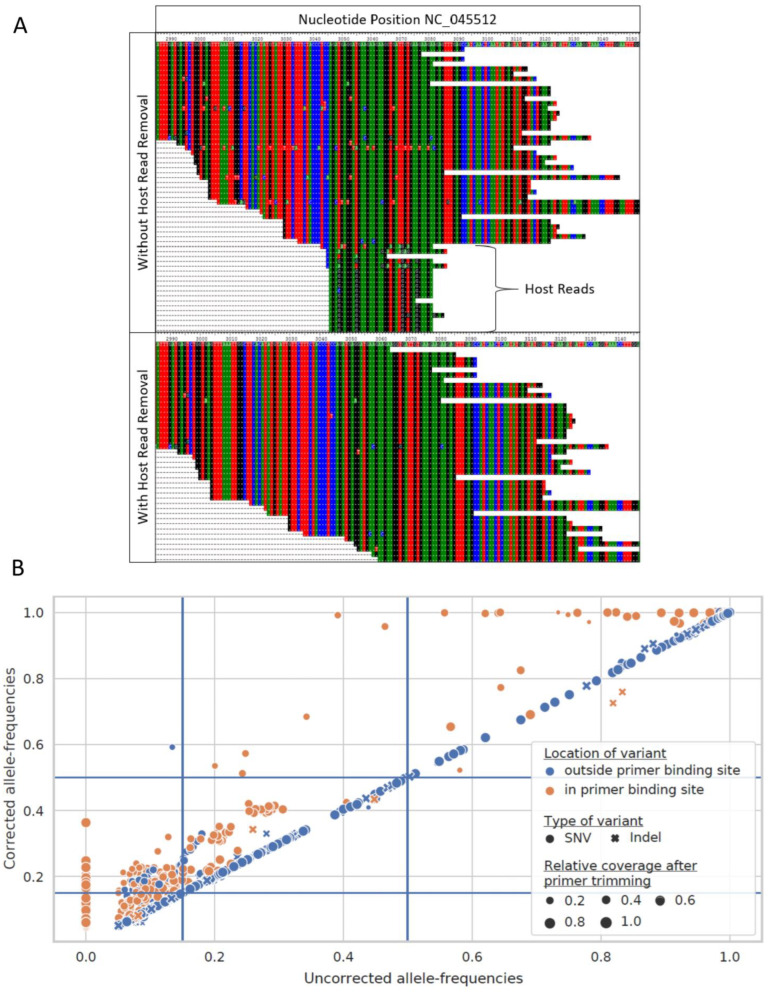
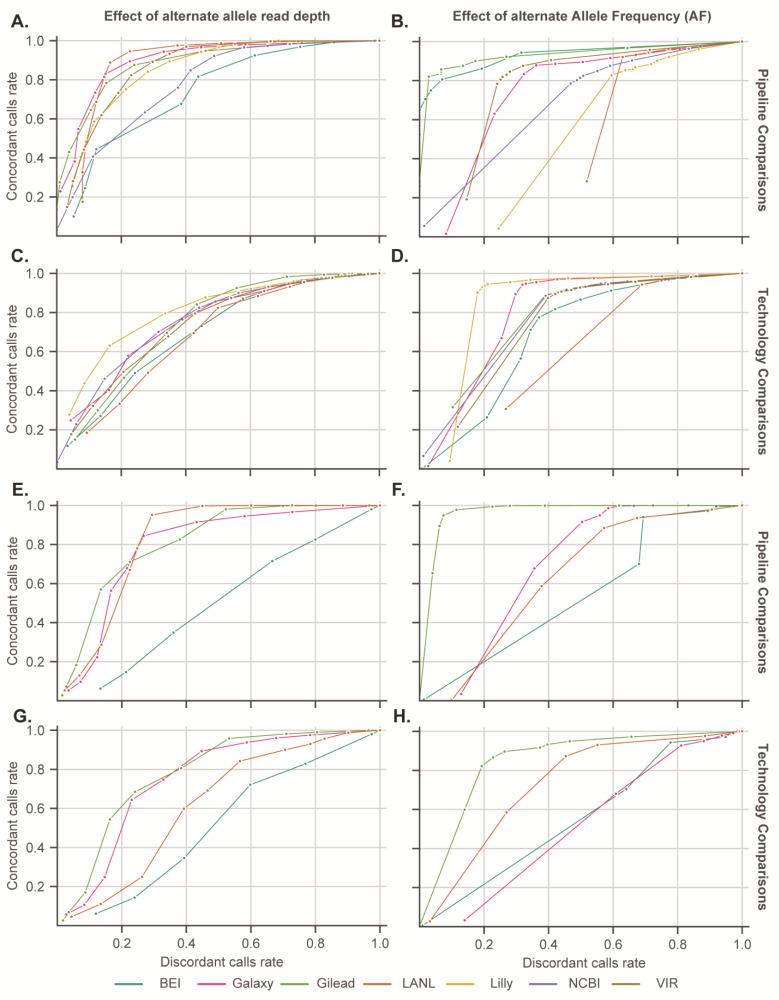
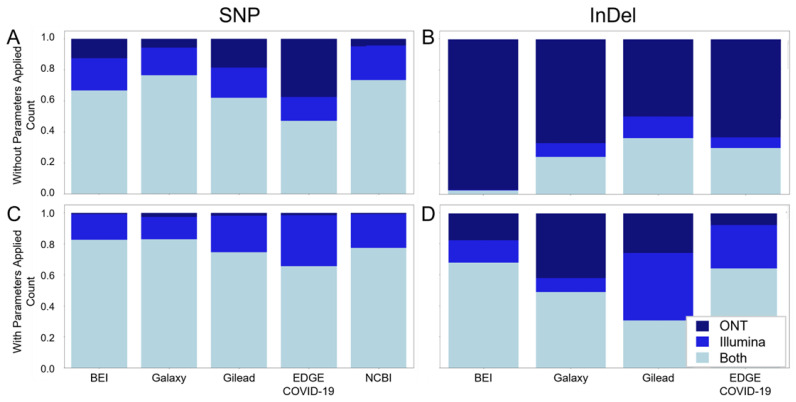
Genomic sequencing of clinical samples to identify emerging variants of SARS-CoV-2 has been a key public health tool for curbing the spread of the virus. As a result, an unprecedented number of SARS-CoV-2 genomes were sequenced during the COVID-19 pandemic, which allowed for rapid identification of genetic variants, enabling the timely design and testing of therapies and deployment of new vaccine formulations to combat the new variants. However, despite the technological advances of deep sequencing, the analysis of the raw sequence data generated globally is neither standardized nor consistent, leading to vastly disparate sequences that may impact identification of variants. Here, we show that for both Illumina and Oxford Nanopore sequencing platforms, downstream bioinformatic protocols used by industry, government, and academic groups resulted in different virus sequences from same sample. These bioinformatic workflows produced consensus genomes with differences in single nucleotide polymorphisms, inclusion and exclusion of insertions, and/or deletions, despite using the same raw sequence as input datasets. Here, we compared and characterized such discrepancies and propose a specific suite of parameters and protocols that should be adopted across the field. Consistent results from bioinformatic workflows are fundamental to SARS-CoV-2 and future pathogen surveillance efforts, including pandemic preparation, to allow for a data-driven and timely public health response.
对临床样本进行基因组测序以鉴定 SARS-CoV-2 的新兴变体一直是遏制病毒传播的主要公共卫生工具。因此,在 COVID-19 大流行期间对 SARS-CoV-2 基因组进行了前所未有的测序,这使得能够快速鉴定遗传变异体,从而及时设计和测试疗法并部署新的疫苗配方来对抗新的变异体。然而,尽管深度测序技术取得了进步,但全球生成的原始序列数据的分析既没有标准化,也不一致,导致差异很大的序列可能会影响变异体的鉴定。在这里,我们表明,对于 Illumina 和 Oxford Nanopore 测序平台,行业、政府和学术团体使用的下游生物信息学协议导致来自同一样本的病毒序列不同。尽管使用相同的原始序列作为输入数据集,但这些生物信息学工作流程生成的共识基因组在单核苷酸多态性、插入的包含和排除以及/或缺失方面存在差异。在这里,我们比较和描述了这些差异,并提出了一套应在整个领域采用的特定参数和协议。生物信息学工作流程的一致结果对于 SARS-CoV-2 和未来的病原体监测工作至关重要,包括大流行准备,以实现数据驱动和及时的公共卫生应对。