School of Pharmacy, Medical Biology Centre, Queen's University Belfast, Belfast, Northern IrelandBT9 7BL, U.K.
J Chem Inf Model. 2022 Oct 10;62(19):4736-4747. doi: 10.1021/acs.jcim.2c00788. Epub 2022 Sep 30.
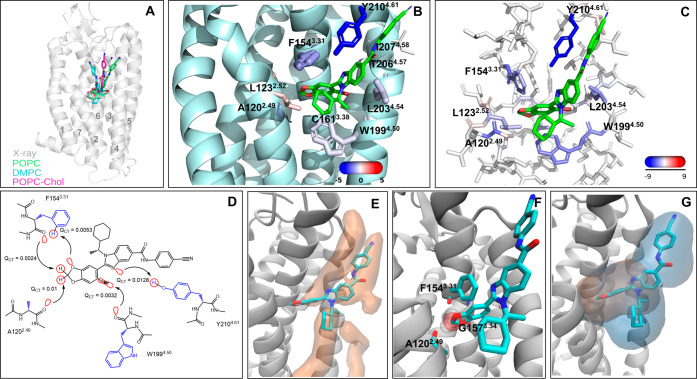
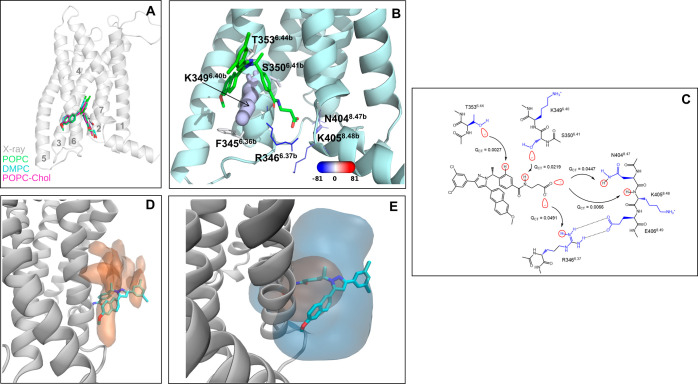
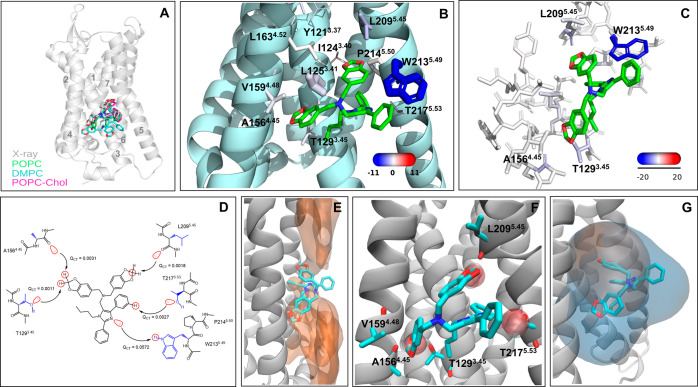
Allosteric modulators are called promising candidates in G protein-coupled receptor (GPCR) drug development by displaying subtype selectivity and more specific receptor modulation. Among the allosteric sites known to date, cavities at the receptor-lipid interface represent an uncharacteristic binding location that raises many questions about the ligand interactions and stability, the binding site structure, and how all of these are affected by lipid molecules. In this work, we analyze interactions in the allosteric sites of the PAR2, C5aR1, and GCGR receptors in three lipid compositions using molecular dynamics simulations. In addition, we performed quantum chemical calculations involving the symmetry-adapted perturbation theory (SAPT) and the natural population analysis to quantify the strength of intermolecular interactions. We show that besides classical hydrogen bonds, weak polar interactions such as O-HC, O-Br, and long-range electrostatics with the backbone amides contribute to the stability of allosteric modulators at the receptor-lipid interface. The allosteric cavities are detectable in various membrane compositions. The availability of polar atoms for interactions in such cavities can be assessed by water molecules from simulations. Although ligand-lipid interactions are weak, lipid tails play a role in ligand binding pose stability and the size of allosteric cavities. We discuss physicochemical aspects of ligand binding at the receptor-lipid interface and suggest a compound library enriched by weak donor groups for ligand search in such sites.
变构调节剂通过显示亚型选择性和更特异性的受体调节,被称为 G 蛋白偶联受体 (GPCR) 药物开发中有前途的候选物。在迄今为止已知的变构位点中,位于受体-脂质界面的腔是一个非典型的结合位置,这引发了许多关于配体相互作用和稳定性、结合位点结构以及所有这些如何受脂质分子影响的问题。在这项工作中,我们使用分子动力学模拟分析了 PAR2、C5aR1 和 GCGR 受体在三种脂质组成中的变构位点的相互作用。此外,我们进行了涉及对称自适应微扰理论 (SAPT) 和自然布居分析的量子化学计算,以量化分子间相互作用的强度。我们表明,除了经典氢键外,弱极性相互作用,如 O-HC、O-Br 和与骨干酰胺的远程静电相互作用,有助于变构调节剂在受体-脂质界面的稳定性。变构腔在各种膜组成中都可检测到。通过模拟中的水分子可以评估此类腔中用于相互作用的极性原子的可用性。尽管配体-脂质相互作用较弱,但脂质尾巴在配体结合构象稳定性和变构腔的大小方面发挥作用。我们讨论了配体在受体-脂质界面结合的物理化学方面,并建议通过弱供体基团丰富配体搜索的化合物库,以在这些位点中进行搜索。