Institute of Metabolic Disease, Baylor Scott and White Research Institute, Dallas, TX 75246, USA.
Department of Medicine, University of Cambridge and Addenbrooke's Hospital, Cambridge CB2 0QQ, UK.
Brain. 2023 Feb 13;146(2):461-474. doi: 10.1093/brain/awac379.
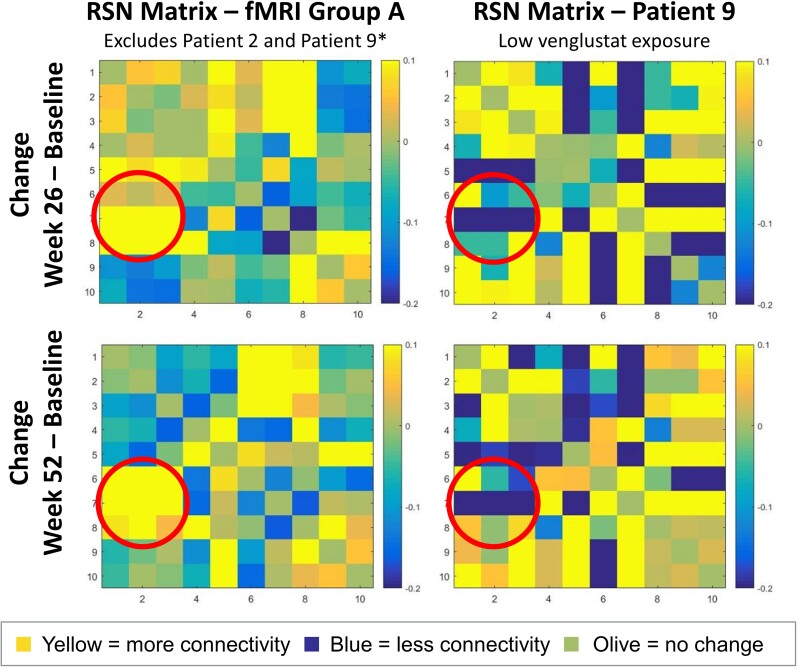
Gaucher disease type 3 is a chronic neuronopathic disorder with wide-ranging effects, including hepatosplenomegaly, anaemia, thrombocytopenia, skeletal disease and diverse neurological manifestations. Biallelic mutations in GBA1 reduce lysosomal acid β-glucosidase activity, and its substrates, glucosylceramide and glucosylsphingosine, accumulate. Enzyme replacement therapy and substrate reduction therapy ameliorate systemic features of Gaucher disease, but no therapies are approved for neurological manifestations. Venglustat is an investigational, brain-penetrant, glucosylceramide synthase inhibitor with potential to improve the disease by rebalancing influx of glucosylceramide with impaired lysosomal recycling. The Phase 2, open-label LEAP trial (NCT02843035) evaluated orally administered venglustat 15 mg once-daily in combination with maintenance dose of imiglucerase enzyme replacement therapy during 1 year of treatment in 11 adults with Gaucher disease type 3. Primary endpoints were venglustat safety and tolerability and change in concentration of glucosylceramide and glucosylsphingosine in CSF from baseline to Weeks 26 and 52. Secondary endpoints included change in plasma concentrations of glucosylceramide and glucosylsphingosine, venglustat pharmacokinetics in plasma and CSF, neurologic function, infiltrative lung disease and systemic disease parameters. Exploratory endpoints included changes in brain volume assessed with volumetric MRI using tensor-based morphometry, and resting functional MRI analysis of regional brain activity and connectivity between resting state networks. Mean (SD) plasma venglustat AUC0-24 on Day 1 was 851 (282) ng•h/ml; Cmax of 58.1 (26.4) ng/ml was achieved at a median tmax 2.00 h. After once-daily venglustat, plasma concentrations (4 h post-dose) were higher compared with Day 1, indicating ∼2-fold accumulation. One participant (Patient 9) had low-to-undetectable venglustat exposure at Weeks 26 and 52. Based on mean plasma and CSF venglustat concentrations (excluding Patient 9), steady state appeared to be reached on or before Week 4. Mean (SD) venglustat concentration at Week 52 was 114 (65.8) ng/ml in plasma and 6.14 (3.44) ng/ml in CSF. After 1 year of treatment, median (inter-quartile range) glucosylceramide decreased 78% (72, 84) in plasma and 81% (77, 83) in CSF; median (inter-quartile range) glucosylsphingosine decreased 56% (41, 60) in plasma and 70% (46, 76) in CSF. Ataxia improved slightly in nine patients: mean (SD, range) total modified Scale for Assessment and Rating of Ataxia score decreased from 2.68 [1.54 (0.0 to 5.5)] at baseline to 1.55 [1.88 (0.0 to 5.0)] at Week 52 [mean change: -1.14 (95% CI: -2.06 to -0.21)]. Whole brain volume increased slightly in patients with venglustat exposure and biomarker reduction in CSF (306.7 ± 4253.3 mm3) and declined markedly in Patient 9 (-13894.8 mm3). Functional MRI indicated stronger connectivity at Weeks 26 and 52 relative to baseline between a broadly distributed set of brain regions in patients with venglustat exposure and biomarker reduction but not Patient 9, although neurocognition, assessed by Vineland II, deteriorated in all domains over time, which illustrates disease progression despite the intervention. There were no deaths, serious adverse events or discontinuations. In adults with Gaucher disease type 3 receiving imiglucerase, addition of once-daily venglustat showed acceptable safety and tolerability and preliminary evidence of clinical stability with intriguing but intrinsically inconsistent signals in selected biomarkers, which need to be validated and confirmed in future research.
戈谢病 3 型是一种慢性神经元疾病,具有广泛的影响,包括肝脾肿大、贫血、血小板减少、骨骼疾病和多种神经表现。GBA1 的双等位基因突变会降低溶酶体酸性β-葡萄糖苷酶的活性,其底物葡萄糖脑苷脂和葡萄糖鞘氨醇会积累。酶替代疗法和底物减少疗法改善了戈谢病的全身特征,但没有针对神经表现的疗法。万格列净是一种具有脑穿透性的葡萄糖脑苷脂合酶抑制剂,通过重新平衡葡萄糖脑苷脂的流入与受损的溶酶体回收,可以改善疾病。这项 2 期、开放性 LEAP 试验(NCT02843035)评估了戈谢病 3 型 11 名成人患者在 1 年的治疗中每天口服 15 毫克万格列净,同时联合维持剂量的伊米苷酶酶替代治疗。主要终点是万格列净的安全性和耐受性,以及从基线到第 26 周和第 52 周脑脊液中葡萄糖脑苷脂和葡萄糖鞘氨醇的浓度变化。次要终点包括血浆中葡萄糖脑苷脂和葡萄糖鞘氨醇的浓度变化、万格列净在血浆和脑脊液中的药代动力学、神经功能、浸润性肺病和全身疾病参数。探索性终点包括使用基于张量的形态测量法评估脑容积的变化,以及使用静息功能磁共振成像分析静息状态网络之间的区域脑活动和连接性的变化。第 1 天的平均(SD)血浆万格列净 AUC0-24 为 851(282)ng•h/ml;中位 tmax 为 2.00 h 时达到 58.1(26.4)ng/ml 的 Cmax。每天一次给予万格列净后,与第 1 天相比,血浆浓度(给药后 4 小时)更高,表明大约有 2 倍的蓄积。一名患者(患者 9)在第 26 周和第 52 周时万格列净的暴露量低至无法检测。基于平均血浆和脑脊液万格列净浓度(排除患者 9),在第 4 周或之前似乎达到了稳态。第 52 周时,血浆(65.8)ng/ml 和脑脊液(3.44)ng/ml 的平均(SD)万格列净浓度分别为 114(65.8)ng/ml 和 6.14(3.44)ng/ml。治疗 1 年后,中位数(四分位间距)血浆葡萄糖脑苷脂降低 78%(72,84),脑脊液降低 81%(77,83);中位数(四分位间距)血浆葡萄糖鞘氨醇降低 56%(41,60),脑脊液降低 70%(46,76)。在 9 名患者中,共济失调略有改善:总改良共济失调评分量表(modified Scale for Assessment and Rating of Ataxia score)的平均(SD,范围)从基线时的 2.68(1.54(0.0 至 5.5))降至 52 周时的 1.55(1.88(0.0 至 5.0))[平均变化:-1.14(95%CI:-2.06 至-0.21)]。有万格列净暴露和脑脊液生物标志物降低的患者脑容量略有增加(306.7±4253.3 mm3),而患者 9 的脑容量显著减少(-13894.8 mm3)。功能磁共振成像显示,与基线相比,在有万格列净暴露和生物标志物降低的患者中,广泛分布的一组脑区之间的连接在第 26 周和第 52 周时更强,但患者 9 除外,尽管所有患者的神经认知能力(通过 Vineland II 评估)随着时间的推移在所有领域都有所恶化,这说明了尽管有干预,但疾病仍在进展。没有死亡、严重不良事件或停药。在接受伊米苷酶治疗的戈谢病 3 型成人患者中,每天添加一次万格列净显示出可接受的安全性和耐受性,以及具有临床稳定性的初步证据,在选定的生物标志物中显示出有趣但本质上不一致的信号,这些需要在未来的研究中进一步验证和确认。