Department of Biochemistry and Molecular Genetics, School of Medicine, University of Colorado Anschutz Medical Campus, Aurora, United States.
Medical Scientist Training Program, University of Colorado Anschutz Medical Campus, Aurora, United States.
Elife. 2022 Oct 19;11:e79940. doi: 10.7554/eLife.79940.
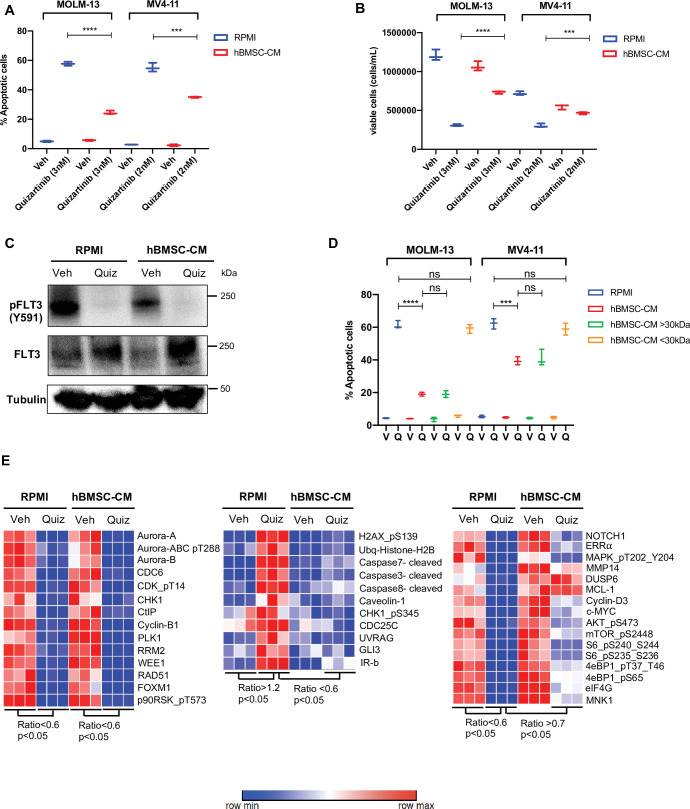
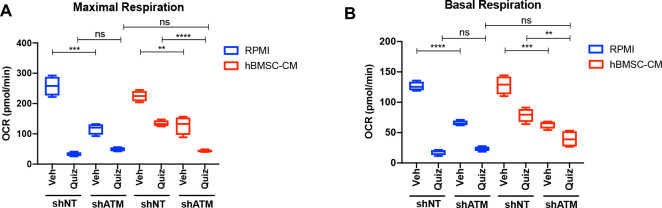
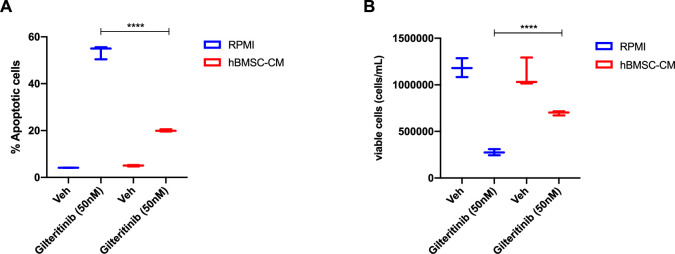
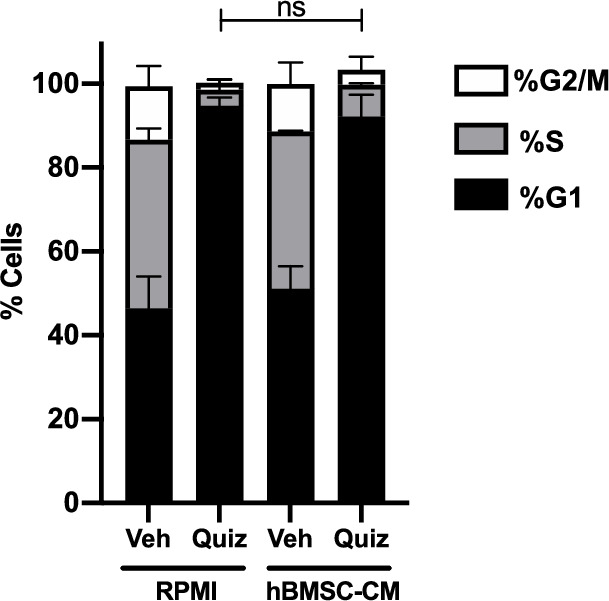
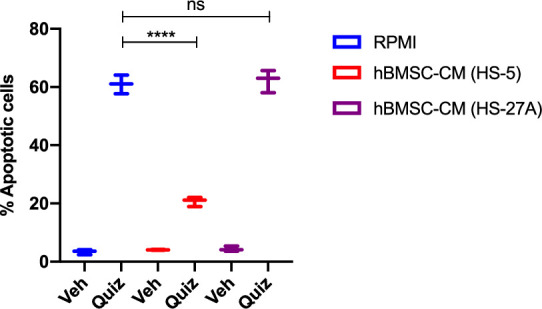
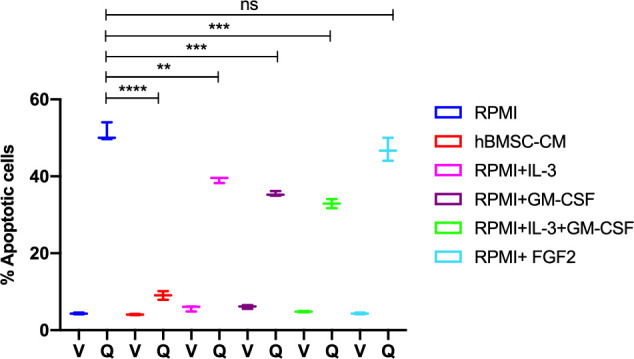
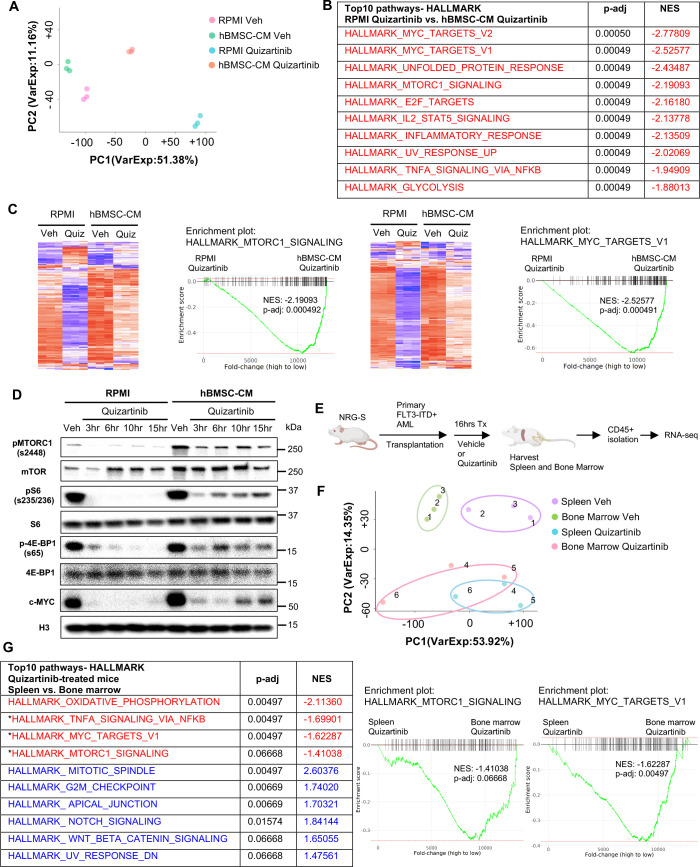
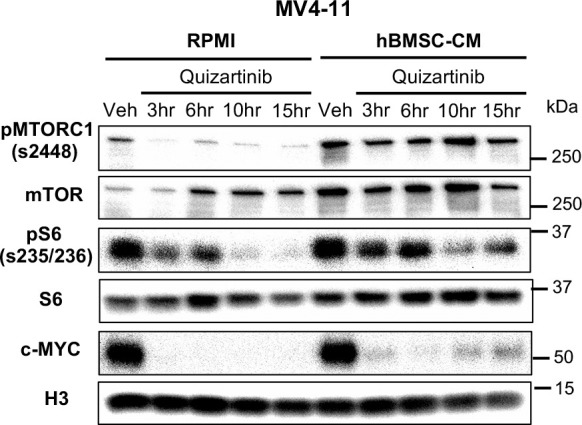
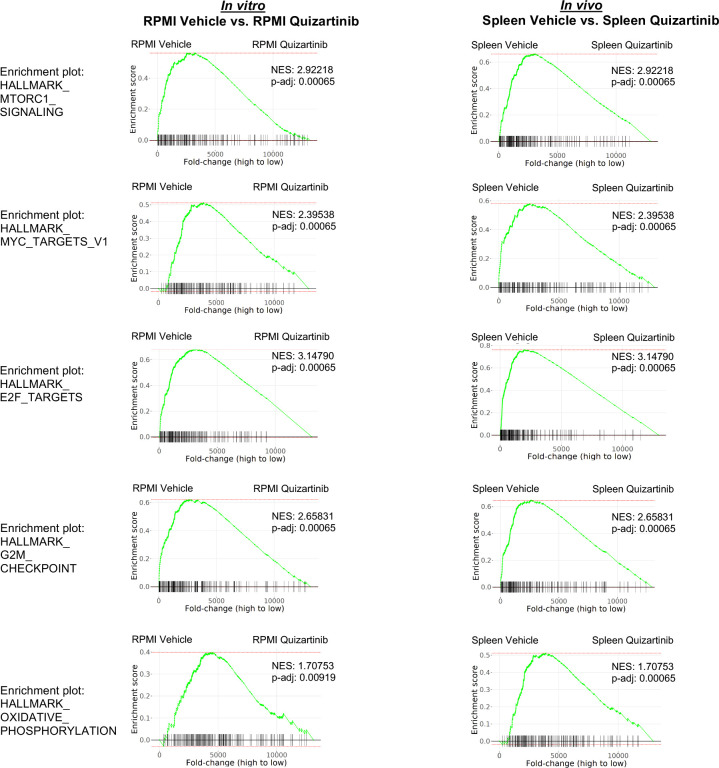
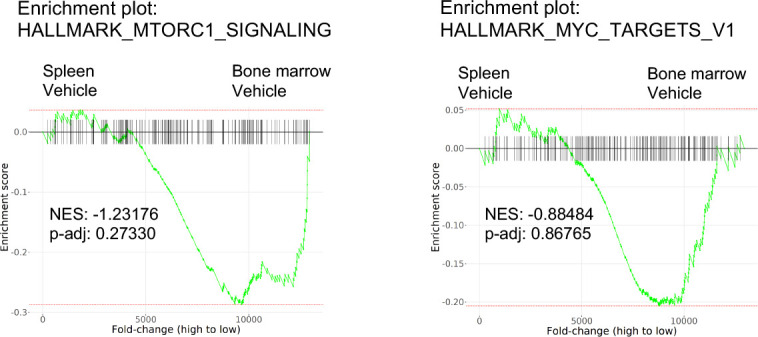
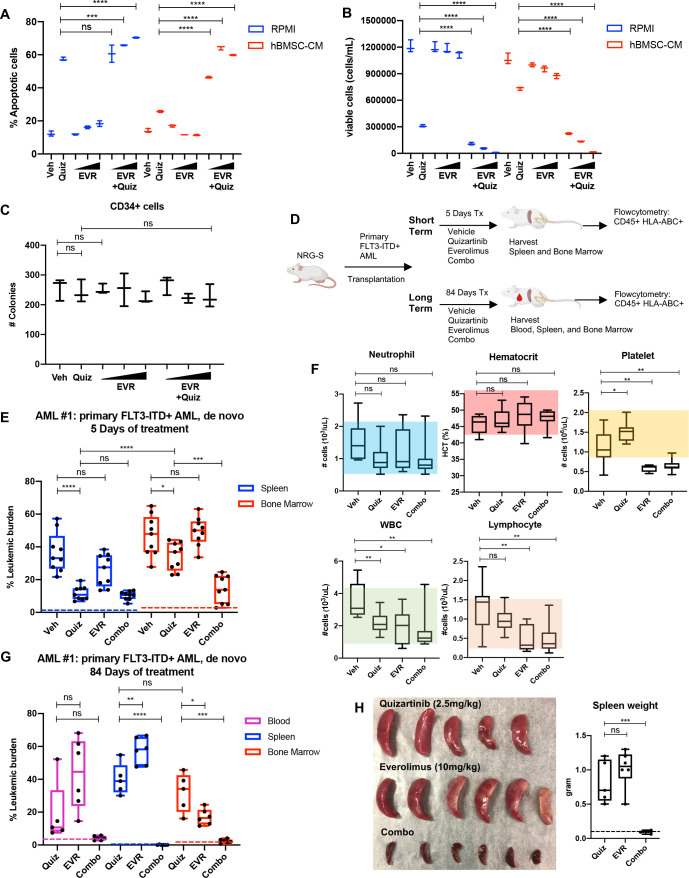
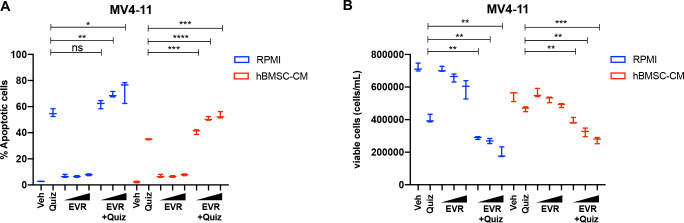
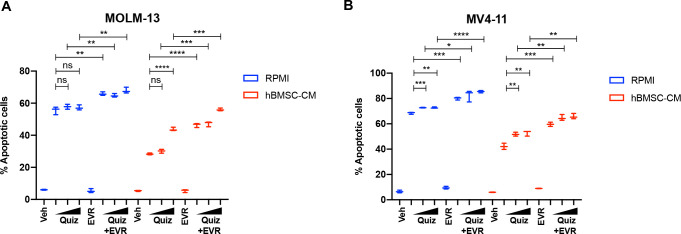
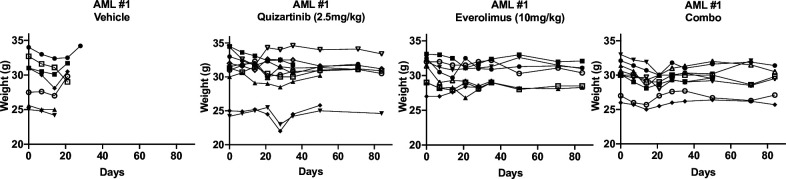
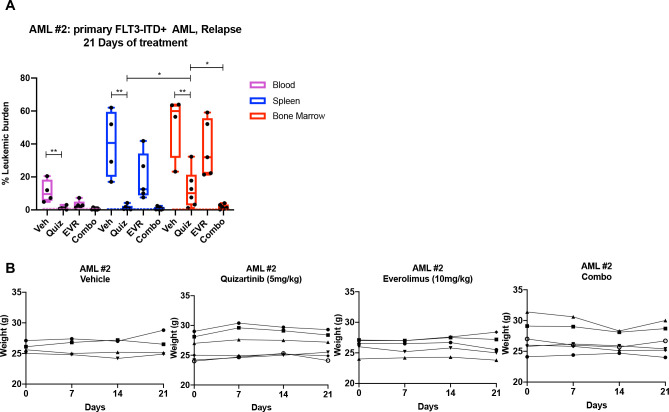
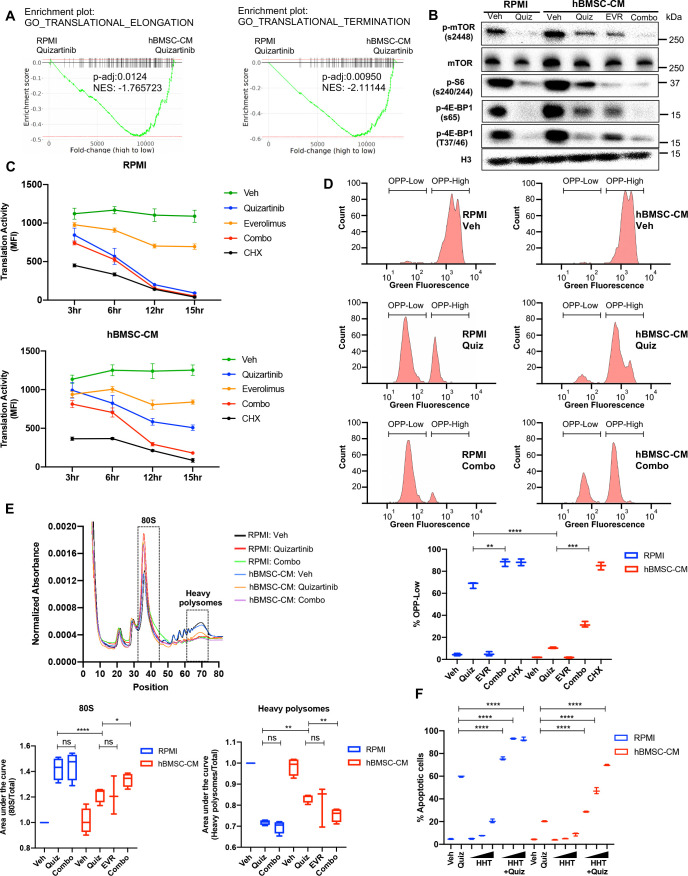
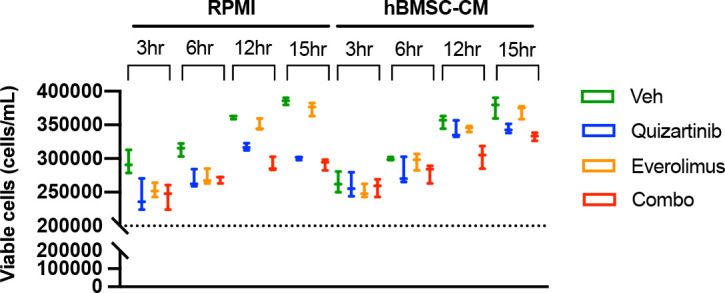
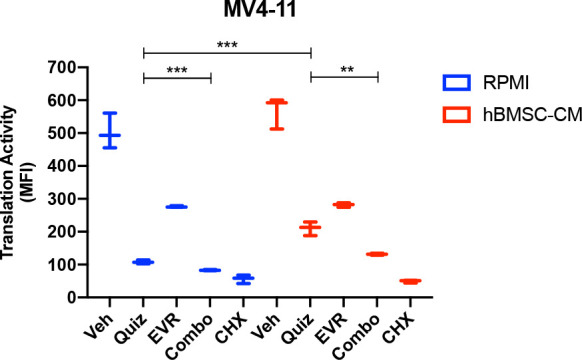
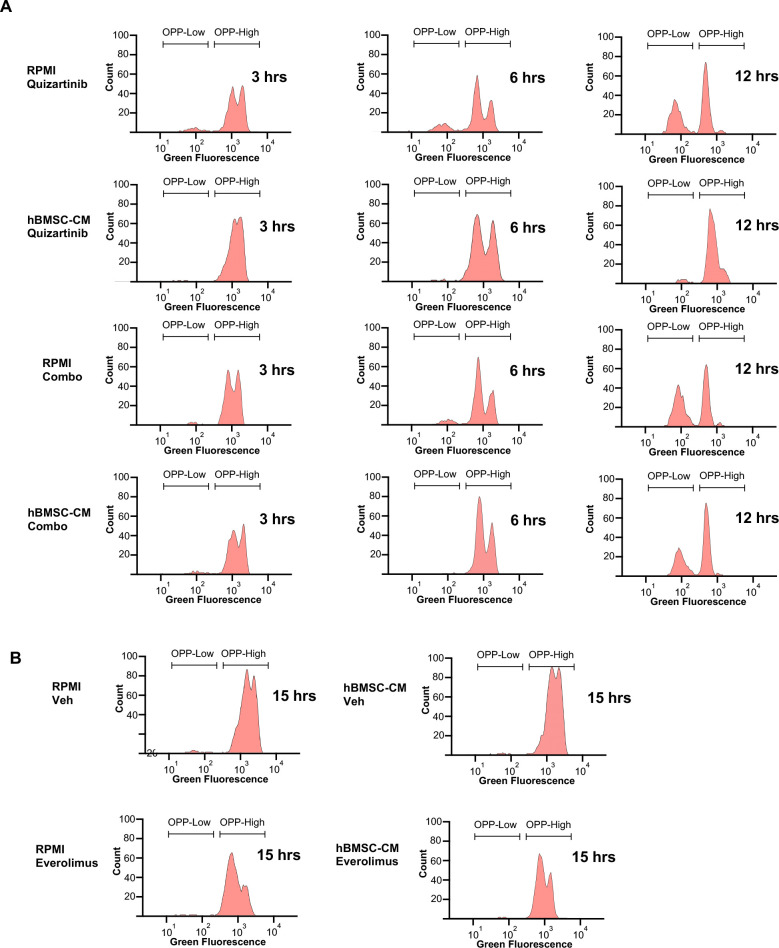
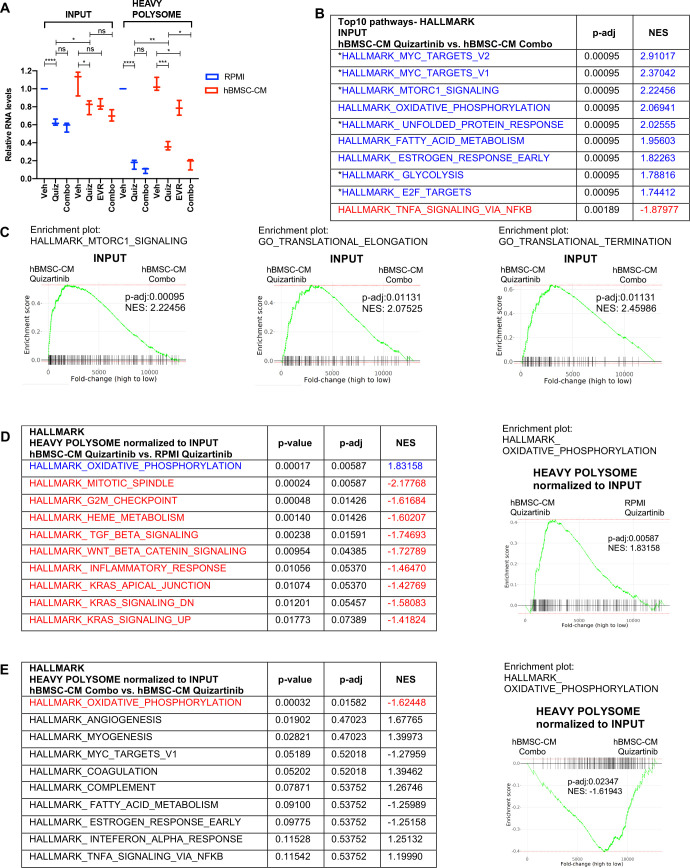
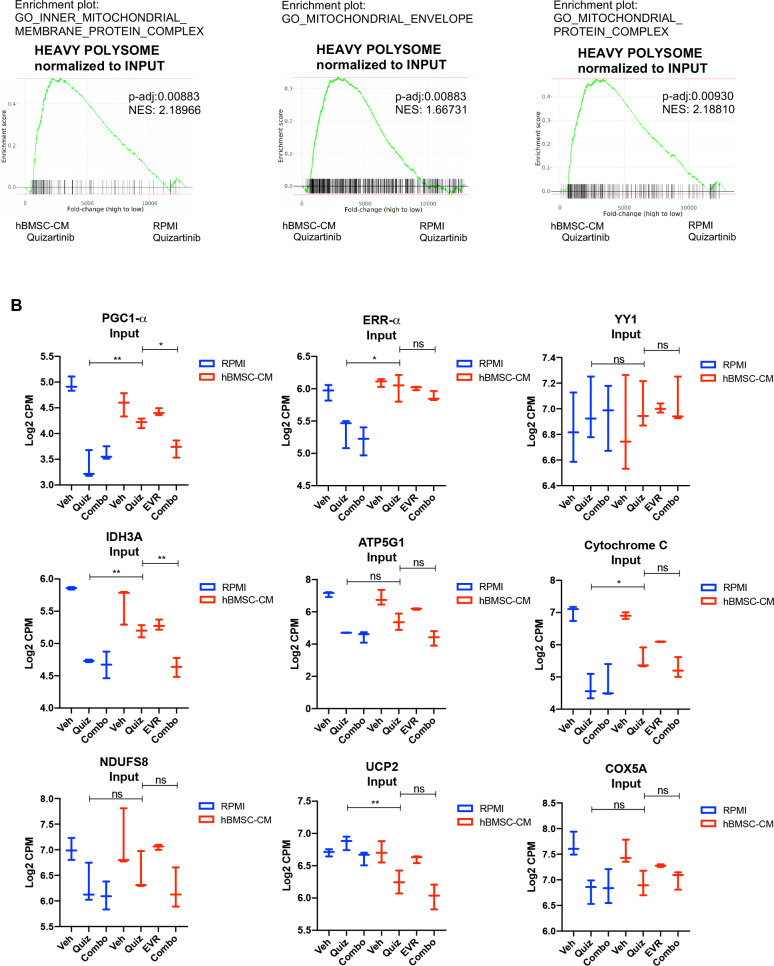
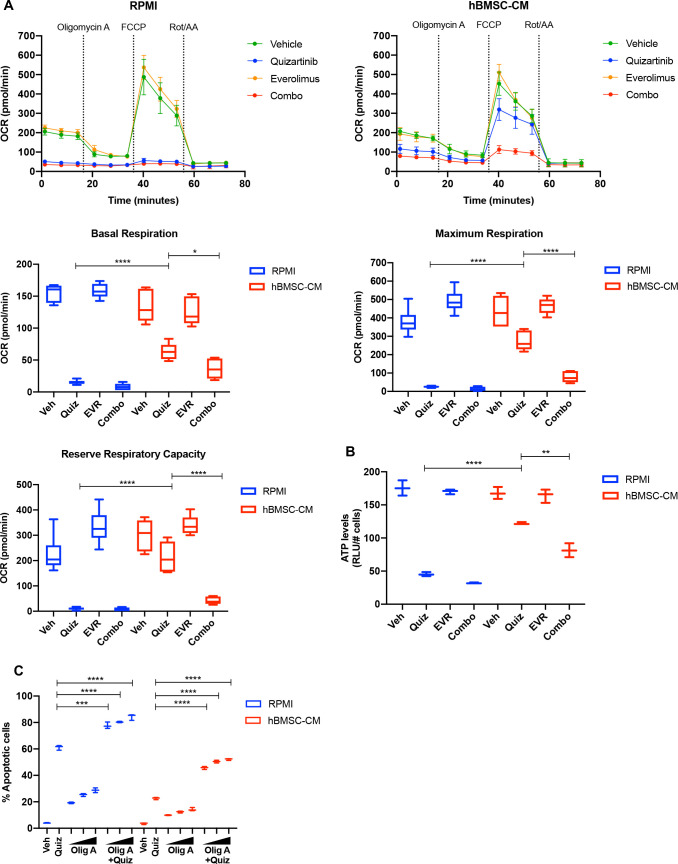
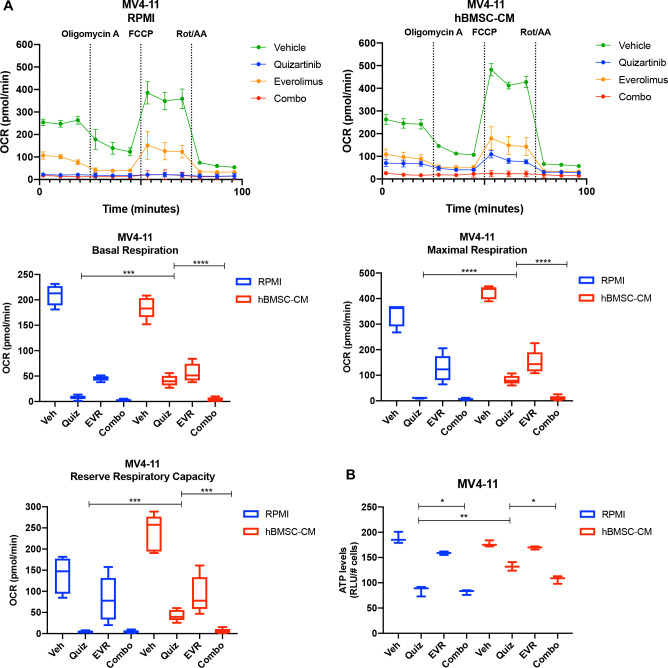
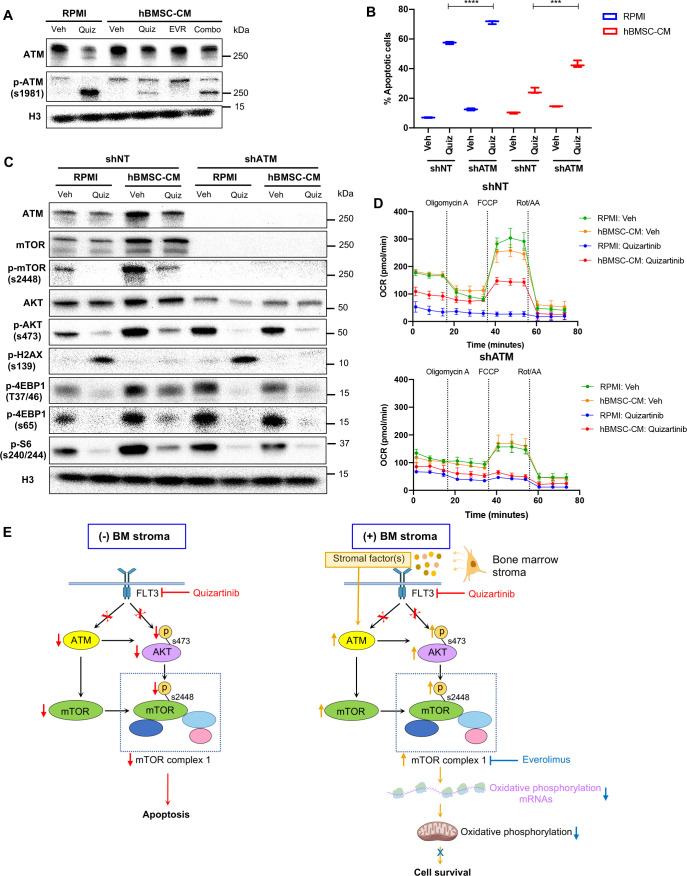
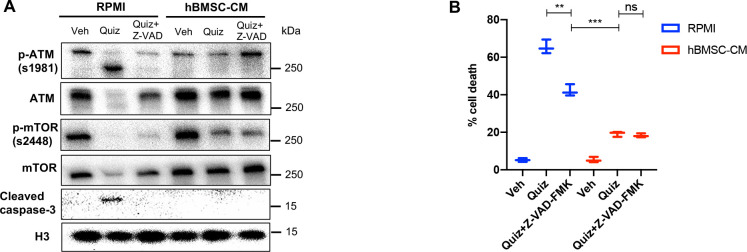
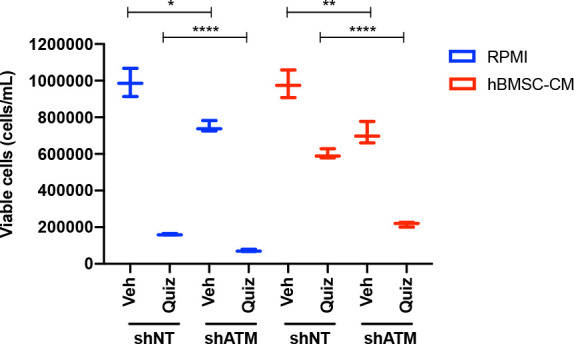
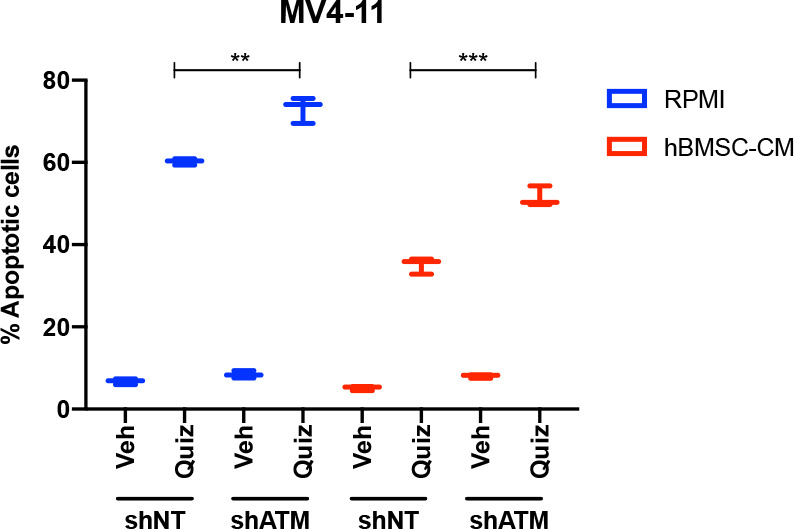
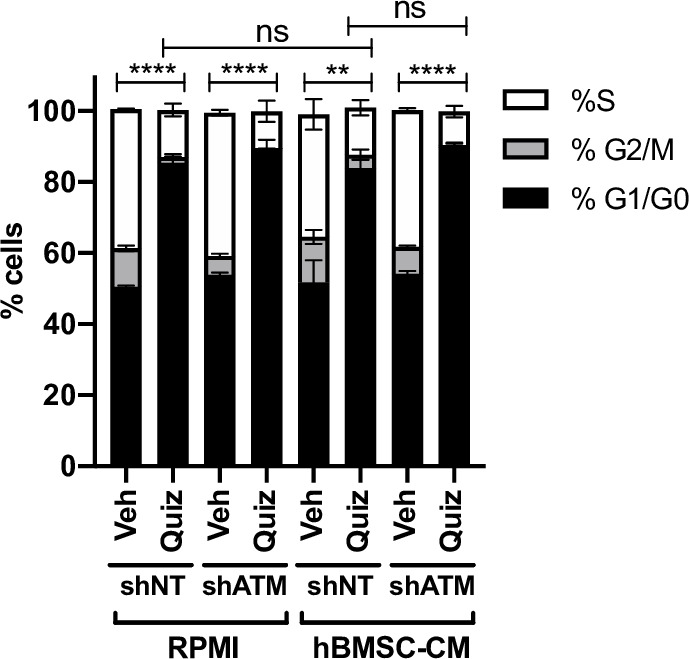
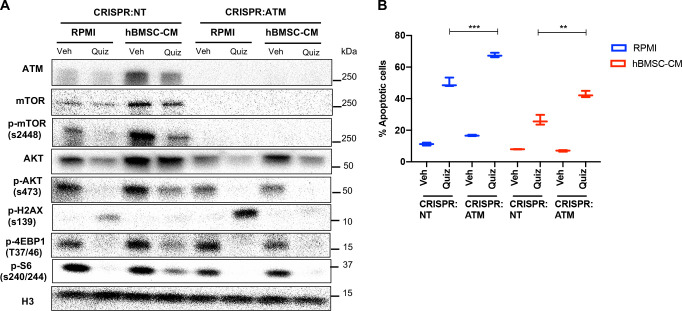
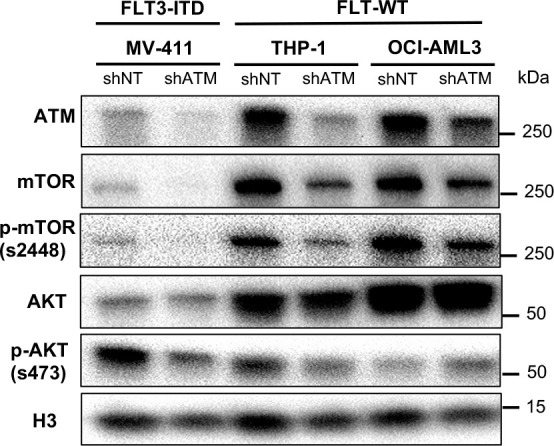
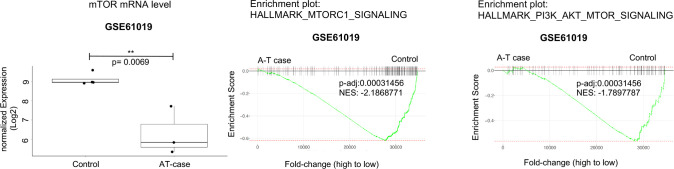
While leukemic cells are susceptible to various therapeutic insults, residence in the bone marrow microenvironment typically confers protection from a wide range of drugs. Thus, understanding the unique molecular changes elicited by the marrow is of critical importance toward improving therapeutic outcomes. In this study, we demonstrate that aberrant activation of oxidative phosphorylation serves to induce therapeutic resistance in FLT3 mutant human AML cells challenged with FLT3 inhibitor drugs. Importantly, our findings show that AML cells are protected from apoptosis following FLT3 inhibition due to marrow-mediated activation of ATM, which in turn upregulates oxidative phosphorylation via mTOR signaling. mTOR is required for the bone marrow stroma-dependent maintenance of protein translation, with selective polysome enrichment of oxidative phosphorylation transcripts, despite FLT3 inhibition. To investigate the therapeutic significance of this finding, we tested the mTOR inhibitor everolimus in combination with the FLT3 inhibitor quizartinib in primary human AML xenograft models. While marrow resident AML cells were highly resistant to quizartinib alone, the addition of everolimus induced profound reduction in tumor burden and prevented relapse. Taken together, these data provide a novel mechanistic understanding of marrow-based therapeutic resistance and a promising strategy for improved treatment of FLT3 mutant AML patients.
虽然白血病细胞容易受到各种治疗性的攻击,但在骨髓微环境中的居留通常可以保护它们免受多种药物的影响。因此,了解骨髓中引起的独特分子变化对于改善治疗效果至关重要。在这项研究中,我们证明了异常激活氧化磷酸化可导致接受 FLT3 抑制剂药物治疗的 FLT3 突变型人类 AML 细胞产生耐药性。重要的是,我们的研究结果表明,AML 细胞在 FLT3 抑制后不会凋亡,这是由于骨髓介导的 ATM 激活所致,而 ATM 又通过 mTOR 信号通路上调氧化磷酸化。mTOR 对于骨髓基质依赖性蛋白质翻译的维持是必需的,尽管 FLT3 被抑制,但氧化磷酸化的转录物仍然选择性地富集在多核糖体上。为了研究这一发现的治疗意义,我们在原发性人类 AML 异种移植模型中测试了 mTOR 抑制剂依维莫司与 FLT3 抑制剂 quizartinib 的联合应用。虽然骨髓驻留的 AML 细胞对 quizartinib 单独治疗具有高度耐药性,但依维莫司的加入可显著降低肿瘤负荷并防止复发。总之,这些数据为骨髓介导的治疗耐药性提供了新的机制理解,并为改善 FLT3 突变型 AML 患者的治疗提供了有希望的策略。