Department of Physiology, School of Medicine, Jinan University, 601 Huangpu Avenue West, Tianhe District, Guangzhou 510632, China.
Center for Clinical Epidemiology and Methodology (CCEM), Guangdong Second Provincial General Hospital, Guangzhou 510317, China.
Cells. 2022 Oct 11;11(20):3184. doi: 10.3390/cells11203184.
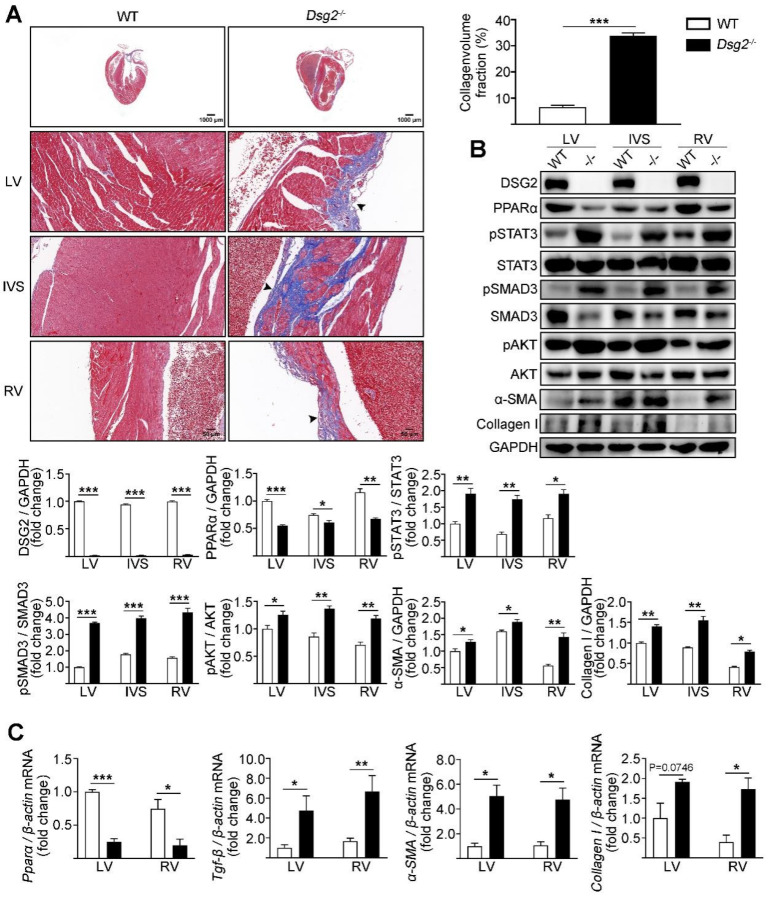
Arrhythmogenic cardiomyopathy (ACM) is a genetic heart muscle disease characterized by progressive fibro-fatty replacement of cardiac myocytes. Up to now, the existing therapeutic modalities for ACM are mostly palliative. About 50% of ACM is caused by mutations in genes encoding desmosomal proteins including Desmoglein-2 (Dsg2). In the current study, the cardiac fibrosis of ACM and its underlying mechanism were investigated by using a cardiac-specific knockout of Dsg2 mouse model.
Cardiac-specific knockout (CS-Dsg2) mice and wild-type (WT) mice were respectively used as the animal model of ACM and controls. The myocardial collagen volume fraction was determined by histological analysis. The expression levels of fibrotic markers such as α-SMA and Collagen I as well as signal transducers such as STAT3, SMAD3, and PPARα were measured by Western blot and quantitative real-time PCR.
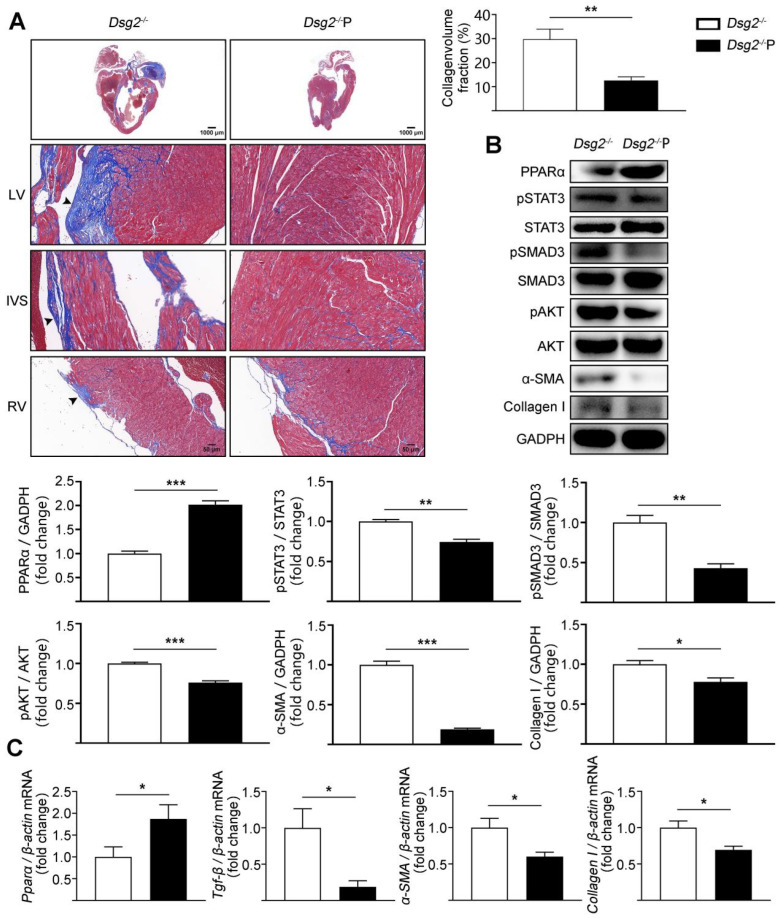
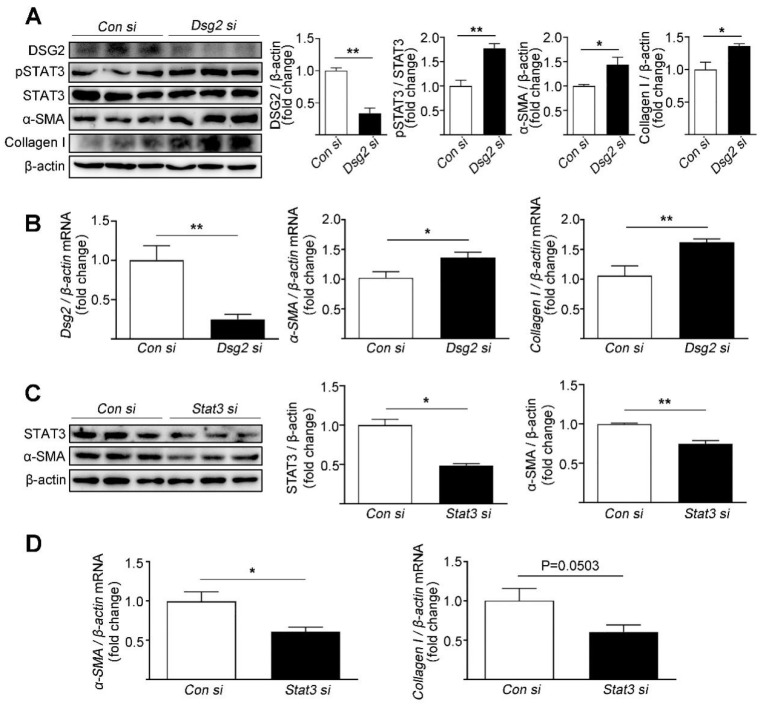
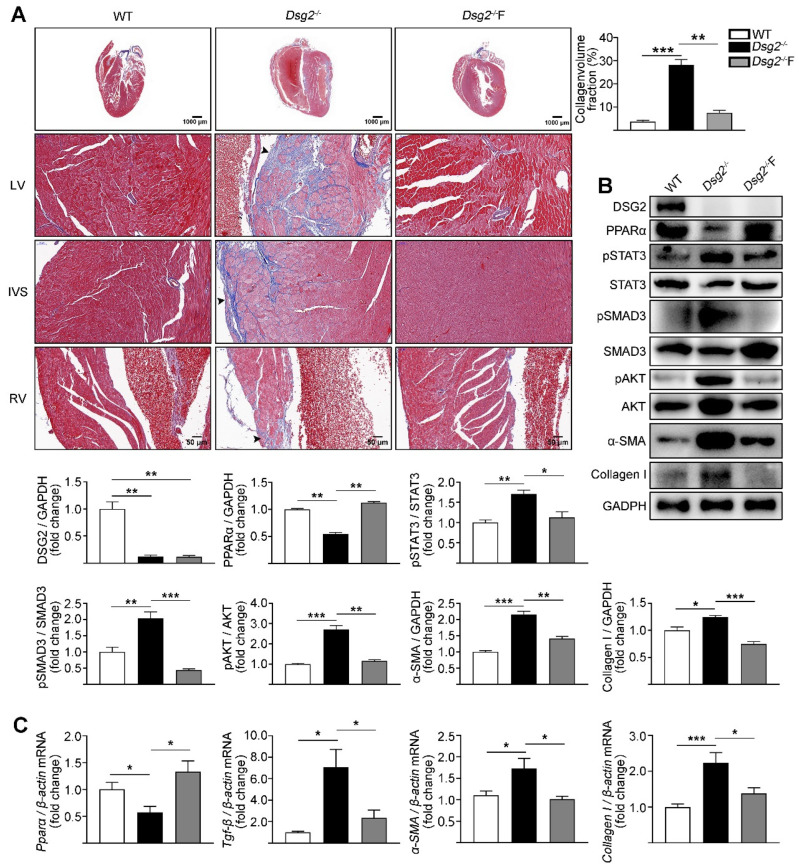
Increased cardiac fibrosis was observed in CS-Dsg2 mice according to Masson staining. PPARα deficiency and hyperactivation of STAT3 and SMAD3 were observed in the myocardium of CS-Dsg2 mice. The biomarkers of fibrosis such as α-SMA and Collagen I were upregulated after gene silencing of Dsg2 in HL-1 cells. Furthermore, STAT3 gene silencing by Stat3 siRNA inhibited the expression of fibrotic markers. The activation of PPARα by fenofibrate or AAV9-Pparα improved the cardiac fibrosis and decreased the phosphorylation of STAT3, SMAD3, and AKT in CS-Dsg2 mice.
Activation of PPARα alleviates the cardiac fibrosis in ACM.
致心律失常性右室心肌病(ACM)是一种遗传性心肌疾病,其特征是心肌细胞进行性纤维脂肪替代。到目前为止,ACM 的现有治疗方法大多是姑息性的。大约 50%的 ACM 是由桥粒蛋白编码基因(包括桥粒芯糖蛋白 2 [Dsg2])的突变引起的。在本研究中,通过使用心脏特异性 Dsg2 敲除小鼠模型研究了 ACM 的心脏纤维化及其潜在机制。
心脏特异性敲除(CS-Dsg2)小鼠和野生型(WT)小鼠分别作为 ACM 的动物模型和对照。通过组织学分析确定心肌胶原容积分数。通过 Western blot 和定量实时 PCR 测量纤维化标志物(如α-SMA 和 Collagen I)和信号转导物(如 STAT3、SMAD3 和 PPARα)的表达水平。
Masson 染色显示 CS-Dsg2 小鼠的心脏纤维化增加。CS-Dsg2 小鼠的心肌中观察到 PPARα 缺失和 STAT3 和 SMAD3 的过度激活。HL-1 细胞中 Dsg2 基因沉默后,纤维化标志物如α-SMA 和 Collagen I 的表达上调。此外,Stat3 siRNA 抑制 STAT3 基因沉默可抑制纤维化标志物的表达。非诺贝特或 AAV9-Pparα 激活 PPARα 可改善 CS-Dsg2 小鼠的心脏纤维化,并降低 CS-Dsg2 小鼠心脏中 STAT3、SMAD3 和 AKT 的磷酸化。
激活 PPARα 可减轻 ACM 中的心脏纤维化。