Department of Education and Support for Regional Medicine, Tohoku University, Seiryo-machi 1-1, Aoba-ku, 980-8574, Sendai, Miyagi, Japan.
COVID-19 Testing Center, Tohoku University, Sendai, Japan.
BMC Ecol Evol. 2022 Oct 28;22(1):123. doi: 10.1186/s12862-022-02078-7.
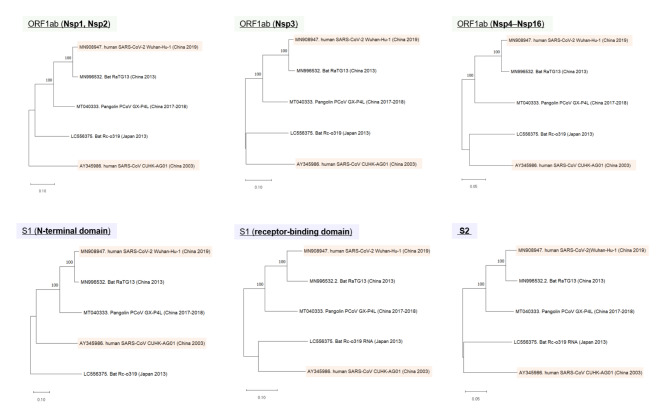
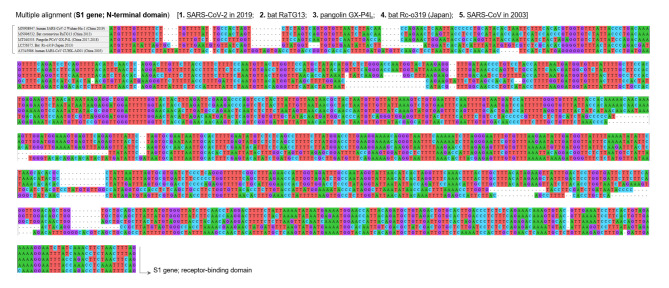
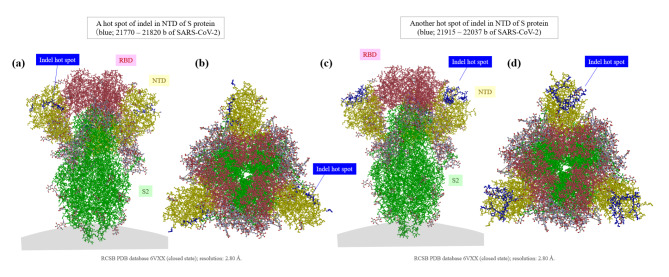
The genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) contains many insertions/deletions (indels) from the genomes of other SARS-related coronaviruses. Some of the identified indels have recently reported to involve relatively long segments of 10-300 consecutive bases and with diverse RNA sequences around gaps between virus species, both of which are different characteristics from the classical shorter in-frame indels. These non-classical complex indels have been identified in non-structural protein 3 (Nsp3), the S1 domain of the spike (S), and open reading frame 8 (ORF8). To determine whether the occurrence of these non-classical indels in specific genomic regions is ubiquitous among broad species of SARS-related coronaviruses in different animal hosts, the present study compared SARS-related coronaviruses from humans (SARS-CoV and SARS-CoV-2), bats (RaTG13 and Rc-o319), and pangolins (GX-P4L), by performing multiple sequence alignment. As a result, indel hotspots with diverse RNA sequences of different lengths between the viruses were confirmed in the Nsp2 gene (approximately 2500-2600 base positions in the overall 29,900 bases), Nsp3 gene (approximately 3000-3300 and 3800-3900 base positions), N-terminal domain of the spike protein (21,500-22,500 base positions), and ORF8 gene (27,800-28,200 base positions). Abnormally high rate of point mutations and complex indels in these regions suggest that the occurrence of mutations in these hotspots may be selectively neutral or even benefit the survival of the viruses. The presence of such indel hotspots has not been reported in different human SARS-CoV-2 strains in the last 2 years, suggesting a lower rate of indels in human SARS-CoV-2. Future studies to elucidate the mechanisms enabling the frequent development of long and complex indels in specific genomic regions of SARS-related coronaviruses would offer deeper insights into the process of viral evolution.
严重急性呼吸综合征冠状病毒 2(SARS-CoV-2)的基因组包含来自其他 SARS 相关冠状病毒基因组的许多插入/缺失(indels)。最近发现的一些插入缺失涉及相对较长的 10-300 个连续碱基,并且在病毒种之间的间隙周围具有不同的 RNA 序列,这两者都与经典的较短框架内插入缺失不同。这些非经典复杂插入缺失已在非结构蛋白 3(Nsp3)、刺突(S)的 S1 结构域和开放阅读框 8(ORF8)中被鉴定出来。为了确定这些非经典插入缺失在不同动物宿主的广泛 SARS 相关冠状病毒中特定基因组区域的发生是否普遍存在,本研究通过进行多序列比对比较了来自人类(SARS-CoV 和 SARS-CoV-2)、蝙蝠(RaTG13 和 Rc-o319)和穿山甲(GX-P4L)的 SARS 相关冠状病毒。结果,在 Nsp2 基因(总体 29900 个碱基中约 2500-2600 个碱基位置)、Nsp3 基因(约 3000-3300 个和 3800-3900 个碱基位置)、刺突蛋白 N 端结构域(21500-22500 个碱基位置)和 ORF8 基因(27800-28200 个碱基位置)中确认了具有不同长度的 RNA 序列的插入缺失热点。这些区域的点突变和复杂插入缺失的异常高发生率表明,这些热点突变的发生可能是选择性中性的,甚至对病毒的存活有益。在过去的 2 年中,不同的人类 SARS-CoV-2 株中没有报道存在这种插入缺失热点,这表明人类 SARS-CoV-2 的插入缺失率较低。未来的研究阐明使 SARS 相关冠状病毒特定基因组区域频繁发生长而复杂插入缺失的机制将为病毒进化过程提供更深入的见解。