Department of Neurology, Medical University of Vienna, Vienna, Austria.
Institute of Human Genetics, Technical University of Munich, Munich, Germany.
J Neurol. 2023 Feb;270(2):909-916. doi: 10.1007/s00415-022-11440-0. Epub 2022 Oct 29.
Congenital myasthenic syndromes (CMS) are a heterogeneous group of disorders caused by genetic defects resulting in impaired neuromuscular transmission. Although effective treatments are available, CMS is probably underdiagnosed, and systematic clinico-genetic investigations are warranted.
We used a nationwide approach to collect Austrian patients with genetically confirmed CMS. We provide a clinical and molecular characterization of this cohort and aimed to ascertain the current frequency of CMS in Austria.
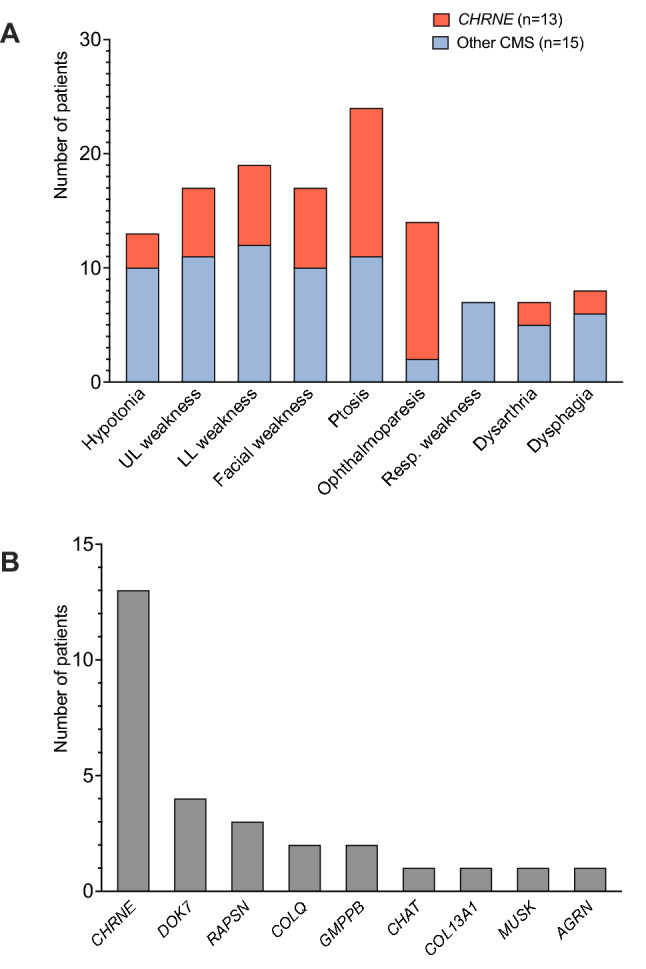
Twenty-eight cases with genetically confirmed CMS were identified, corresponding to an overall prevalence of 3.1 per million (95% CI 2.0-4.3) in Austria. The most frequent genetic etiology was CHRNE (n = 13), accounting for 46.4% of the cohort. Within this subgroup, the variant c.1327del, p.(Glu443Lysfs*64) was detected in nine individuals. Moreover, causative variants were found in DOK7 (n = 4), RAPSN (n = 3), COLQ (n = 2), GMPPB (n = 2), CHAT (n = 1), COL13A1 (n = 1), MUSK (n = 1) and AGRN (n = 1). Clinical onset within the first year of life was reported in one half of the patients. Across all subtypes, the most common symptoms were ptosis (85.7%), lower limb (67.9%), upper limb (60.7%) and facial weakness (60.7%). The majority of patients (96.4%) received specific treatment, including acetylcholinesterase inhibitors in 20, adrenergic agonists in 11 and 3,4-diaminopyridine in nine patients.
Our study presents the first systematic characterization of individuals with CMS in Austria, providing prevalence estimates and genotype-phenotype correlations that may help to improve the diagnostic approach and patient management.
先天性肌无力综合征(CMS)是一组由遗传缺陷引起的异质性疾病,导致神经肌肉传递受损。尽管有有效的治疗方法,但 CMS 可能被低估了,因此需要进行系统的临床遗传研究。
我们采用全国性方法收集了奥地利经基因证实的 CMS 患者。我们对该队列进行了临床和分子特征描述,并旨在确定 CMS 在奥地利的当前频率。
共确定了 28 例经基因证实的 CMS 病例,在奥地利的总体患病率为 3.1/百万人(95%CI 2.0-4.3)。最常见的遗传病因是 CHRNE(n=13),占队列的 46.4%。在这个亚组中,发现了变体 c.1327del,p.(Glu443Lysfs*64),在 9 个人中。此外,在 DOK7(n=4)、RAPSN(n=3)、COLQ(n=2)、GMPPB(n=2)、CHAT(n=1)、COL13A1(n=1)、MUSK(n=1)和 AGRN(n=1)中也发现了致病变体。一半的患者在生命的第一年出现临床发病。在所有亚型中,最常见的症状是上睑下垂(85.7%)、下肢(67.9%)、上肢(60.7%)和面部无力(60.7%)。大多数患者(96.4%)接受了特定治疗,包括乙酰胆碱酯酶抑制剂 20 例,肾上腺素能激动剂 11 例,3,4-二氨基吡啶 9 例。
我们的研究首次对奥地利 CMS 患者进行了系统描述,提供了患病率估计和基因型-表型相关性,这可能有助于改善诊断方法和患者管理。