Division of Clinical Microbiology, Department of Laboratory Medicine and Pathology, Mayo Clinicgrid.66875.3a, Rochester, Minnesota, USA.
Ares Genetics GmbH, Vienna, Austria.
Microbiol Spectr. 2022 Dec 21;10(6):e0392022. doi: 10.1128/spectrum.03920-22. Epub 2022 Nov 9.
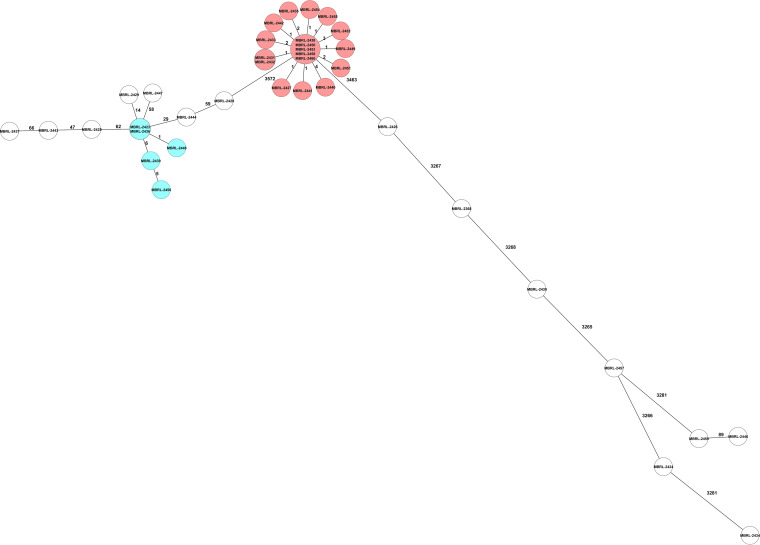
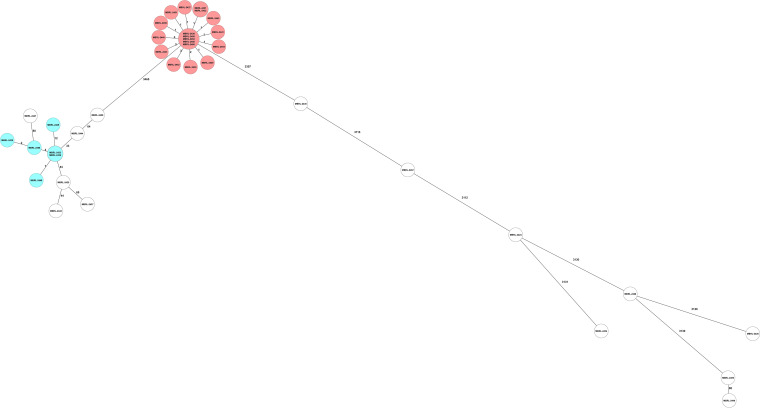
Over the past decade, whole-genome sequencing (WGS) has overtaken traditional bacterial typing methods for studies of genetic relatedness. Further, WGS data generated during epidemiologic studies can be used in other clinically relevant bioinformatic applications, such as antibiotic resistance prediction. Using commercially available software tools, the relatedness of 38 clinical isolates of multidrug-resistant Pseudomonas aeruginosa was defined by two core genome multilocus sequence typing (cgMLST) methods, and the WGS data of each isolate was analyzed to predict antibiotic susceptibility to nine antibacterial agents. The WGS typing and resistance prediction data were compared with pulsed-field gel electrophoresis (PFGE) and phenotypic antibiotic susceptibility results, respectively. Simpson's Diversity Index and adjusted Wallace pairwise assessments of the three typing methods showed nearly identical discriminatory power. Antibiotic resistance prediction using a trained analytical pipeline examined 342 bacterial-drug combinations with an overall categorical agreement of 92.4% and very major, major, and minor error rates of 3.6, 4.1, and 4.1%, respectively. Multidrug-resistant Pseudomonas aeruginosa isolates are a serious public health concern due to their resistance to nearly all or all of the available antibiotics, including carbapenems. Utilizing molecular approaches in conjunction with antibiotic susceptibility prediction software warrants investigation for use in the clinical laboratory workflow. These molecular tools coupled with antibiotic resistance prediction tools offer the opportunity to overcome the extended turnaround time and technical challenges of phenotypic susceptibility testing.
在过去的十年中,全基因组测序(WGS)已经取代了传统的细菌分型方法,用于研究遗传相关性。此外,在流行病学研究中生成的 WGS 数据可用于其他与临床相关的生物信息学应用,例如抗生素耐药性预测。使用商业上可用的软件工具,通过两种核心基因组多位点序列分型(cgMLST)方法定义了 38 株多药耐药铜绿假单胞菌的临床分离株的相关性,并对每个分离株的 WGS 数据进行分析以预测对九种抗菌药物的敏感性。WGS 分型和耐药预测数据分别与脉冲场凝胶电泳(PFGE)和表型药敏结果进行了比较。三种分型方法的 Simpson 多样性指数和调整后的 Wallace 成对评估显示出几乎相同的区分能力。使用经过训练的分析管道对 342 种细菌-药物组合进行抗生素耐药性预测,总体分类一致性为 92.4%,非常主要、主要和次要错误率分别为 3.6%、4.1%和 4.1%。由于几乎对所有或所有可用抗生素(包括碳青霉烯类)都具有耐药性,多药耐药铜绿假单胞菌分离株是一个严重的公共卫生问题。结合抗生素药敏预测软件使用分子方法值得在临床实验室工作流程中进行研究。这些分子工具结合抗生素耐药性预测工具为克服表型药敏试验的延长周转时间和技术挑战提供了机会。