NSF-Simons Center for the Mathematical and Statistical Analysis of Biology, Harvard University, Cambridge, MA 02138, USA.
Department of Mathematics, University of California Berkeley, Berkeley, CA 94720, USA.
Bioinformatics. 2023 Jan 1;39(1). doi: 10.1093/bioinformatics/btac724.
Multiple sequence alignments (MSAs) of homologous sequences contain information on structural and functional constraints and their evolutionary histories. Despite their importance for many downstream tasks, such as structure prediction, MSA generation is often treated as a separate pre-processing step, without any guidance from the application it will be used for.
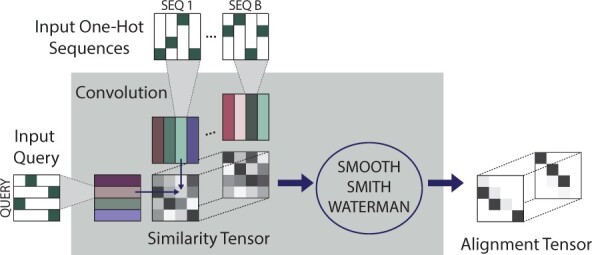
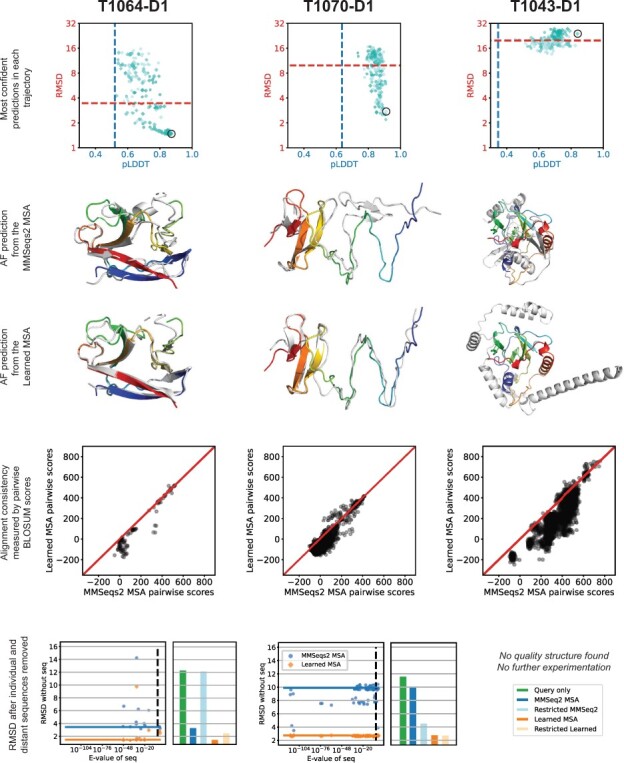
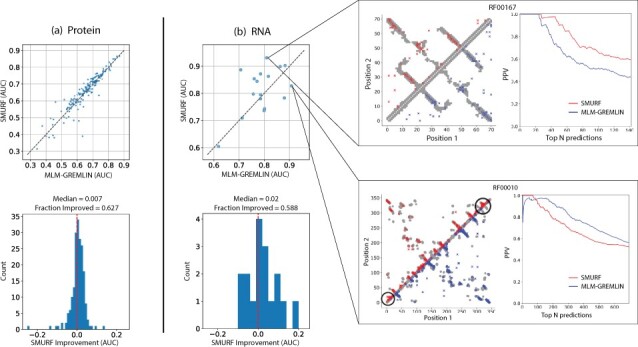
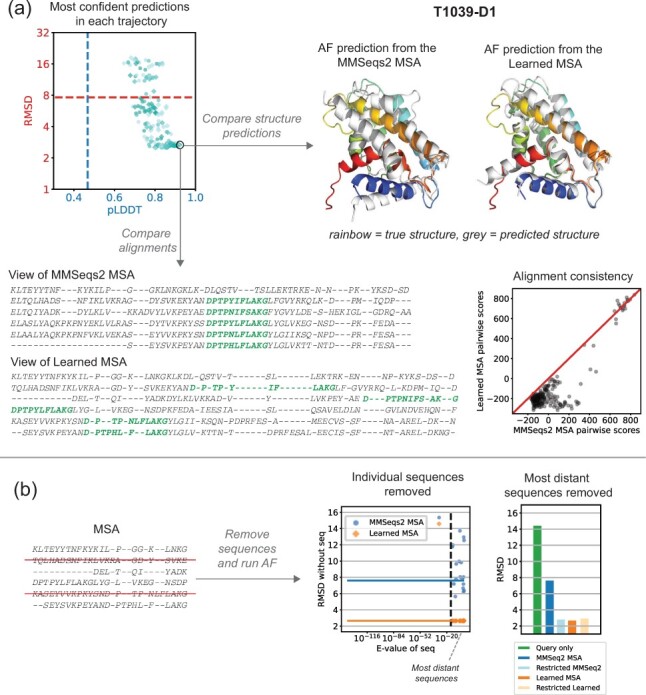
Here, we implement a smooth and differentiable version of the Smith-Waterman pairwise alignment algorithm that enables jointly learning an MSA and a downstream machine learning system in an end-to-end fashion. To demonstrate its utility, we introduce SMURF (Smooth Markov Unaligned Random Field), a new method that jointly learns an alignment and the parameters of a Markov Random Field for unsupervised contact prediction. We find that SMURF learns MSAs that mildly improve contact prediction on a diverse set of protein and RNA families. As a proof of concept, we demonstrate that by connecting our differentiable alignment module to AlphaFold2 and maximizing predicted confidence, we can learn MSAs that improve structure predictions over the initial MSAs. Interestingly, the alignments that improve AlphaFold predictions are self-inconsistent and can be viewed as adversarial. This work highlights the potential of differentiable dynamic programming to improve neural network pipelines that rely on an alignment and the potential dangers of optimizing predictions of protein sequences with methods that are not fully understood.
Our code and examples are available at: https://github.com/spetti/SMURF.
Supplementary data are available at Bioinformatics online.
同源序列的多重序列比对 (MSA) 包含有关结构和功能约束及其进化历史的信息。尽管它们对于许多下游任务(如结构预测)非常重要,但 MSA 的生成通常被视为一个单独的预处理步骤,而没有任何关于其应用的指导。
在这里,我们实现了一种平滑可微的 Smith-Waterman 两两比对算法版本,能够以端到端的方式联合学习 MSA 和下游机器学习系统。为了证明其效用,我们引入了 SMURF(平滑 Markov 未对齐随机场),这是一种新的方法,它联合学习对齐和 Markov 随机场的参数,用于无监督接触预测。我们发现,SMURF 学习的 MSA 可以在各种蛋白质和 RNA 家族上适度提高接触预测的准确性。作为概念验证,我们证明通过将我们的可微分对齐模块连接到 AlphaFold2 并最大化预测置信度,我们可以学习到可以提高初始 MSA 结构预测的 MSA。有趣的是,改善 AlphaFold 预测的比对是自相矛盾的,可以视为对抗性的。这项工作强调了可微分动态规划在改善依赖于比对的神经网络管道方面的潜力,以及使用不完全理解的方法优化蛋白质序列预测的潜在危险。
我们的代码和示例可在以下网址获得:https://github.com/spetti/SMURF。
补充数据可在 Bioinformatics 在线获得。