Department of Natural Resources and Environmental Engineering, Shiraz University, Shiraz, Iran.
School of Biological Sciences, The University of Auckland, Auckland, New Zealand.
BMC Genomics. 2022 Nov 12;23(1):750. doi: 10.1186/s12864-022-08984-w.
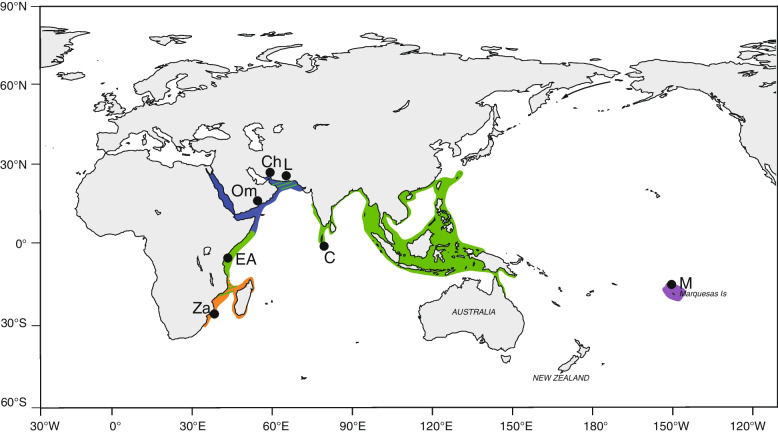
Evolutionary divergence and speciation often occur at a slower rate in the marine realm due to the higher potential for long-distance reproductive interaction through larval dispersal. One common evolutionary pattern in the Indo-Pacific, is divergence of populations and species at the peripheries of widely-distributed organisms. However, the evolutionary and demographic histories of such divergence are yet to be well understood. Here we address these issues by coupling genome-wide SNP data with mitochondrial DNA sequences to test the patterns of genetic divergence and possible secondary contact among geographically distant populations of the highly valuable spiny lobster Panulirus homarus species complex, distributed widely through the Indo-Pacific, from South Africa to the Marquesas Islands.
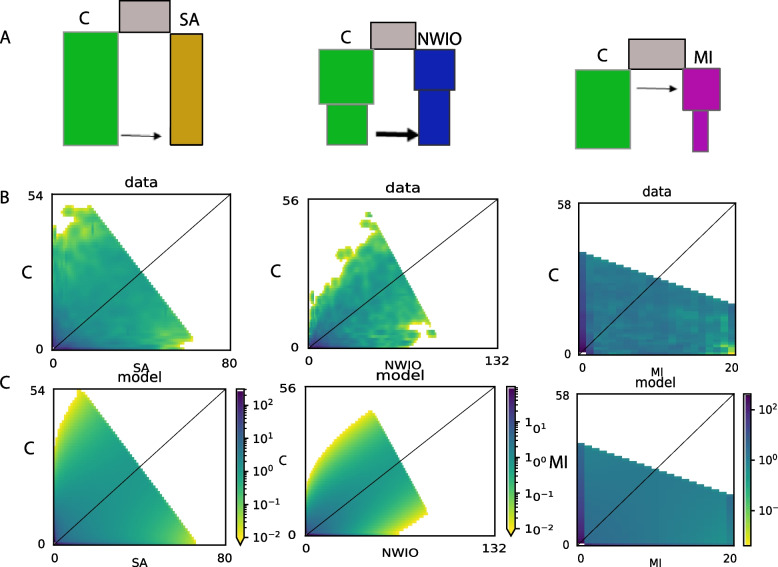
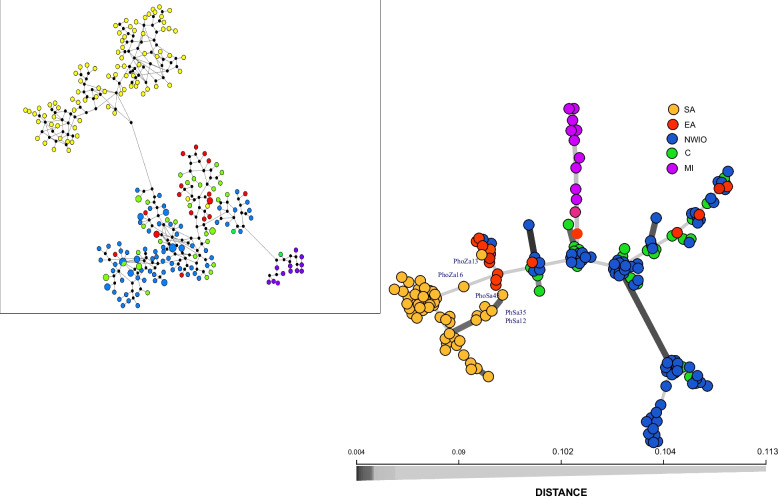
After stringent filtering, 2020 SNPs were used for population genetic and demographic analyses, revealing strong regional structure (F = 0.148, P < 0001), superficially in accordance with previous analyses. However, detailed demographic analyses supported a much more complex evolutionary history of these populations, including a hybrid origin of a North-West Indian Ocean (NWIO) population, which has previously been discriminated morphologically, but not genetically. The best-supported demographic models suggested that the current genetic relationships among populations were due to a complex series of past divergences followed by asymmetric migration in more recent times.
Overall, this study suggests that alternating periods of marine divergence and gene flow have driven the current genetic patterns observed in this lobster and may help explain the observed wider patterns of marine species diversity in the Indo-Pacific.
由于幼虫扩散增加了远距离生殖相互作用的可能性,海洋环境中的进化分歧和物种形成通常比在陆地环境中更为缓慢。在印度-太平洋地区,一个常见的进化模式是广泛分布的生物的种群和物种在边缘的分歧。然而,这种分歧的进化和人口历史尚未得到很好的理解。在这里,我们通过将全基因组 SNP 数据与线粒体 DNA 序列相结合,来解决这些问题,以测试遗传分歧的模式和地理上遥远的种群之间可能的二次接触,这些种群分布广泛,从南非到马克萨斯群岛。
经过严格筛选,使用 2020 个 SNP 进行了种群遗传和人口分析,结果显示出强烈的区域结构(F=0.148,P<0001),表面上与以前的分析一致。然而,详细的人口分析支持了这些种群更为复杂的进化历史,包括西北印度洋(NWIO)种群的杂种起源,该种群以前在形态上有所区分,但在遗传上没有。支持度最高的人口模型表明,目前种群之间的遗传关系是由于过去一系列复杂的分歧,以及最近不对称的迁移造成的。
总的来说,这项研究表明,海洋分歧和基因流动的交替时期推动了目前在这种龙虾中观察到的遗传模式,这可能有助于解释印度-太平洋地区海洋物种多样性的观察到的更广泛模式。