Carvalho Alexandra, Negi Suchit, Neto Antonio H Castro
Institute for Functional Intelligent Materials, National University of Singapore, Singapore, 117544, Singapore.
Centre for Advanced 2D Materials, National University of Singapore, Singapore, 117546, Singapore.
Sci Rep. 2022 Nov 19;12(1):19930. doi: 10.1038/s41598-022-21561-1.
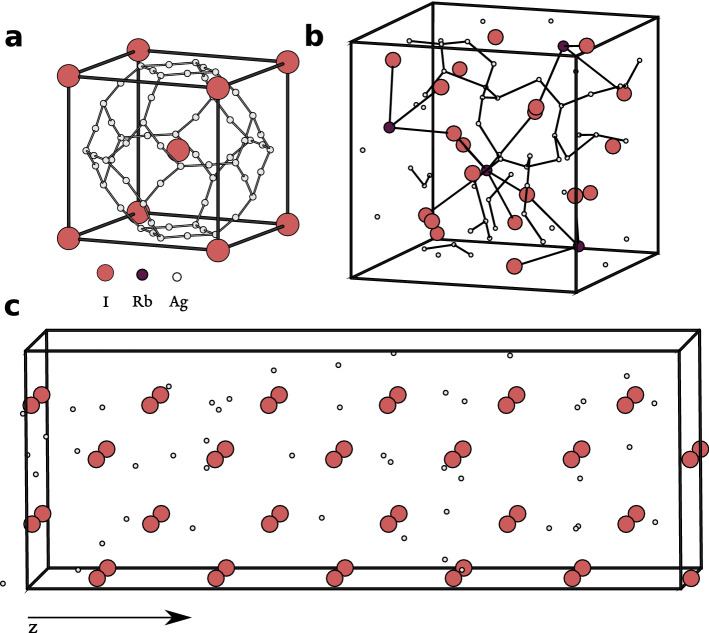
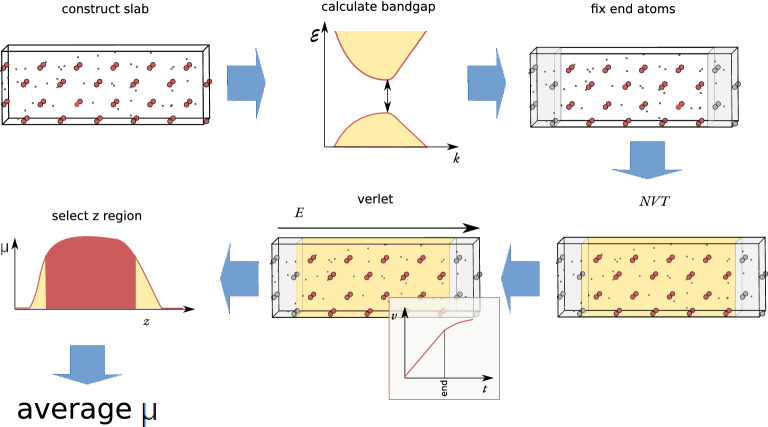
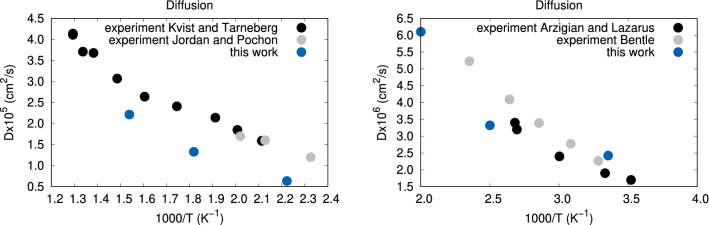
We describe an approach based on non-equilibrium molecular dynamics (NEMD) simulations to calculate the ionic mobility of solid ion conductors such as solid electrolytes from first-principles. The calculations are carried out in finite slabs of the material, where an electric field is applied and the dynamic response of the mobile ions is measured. We compare our results with those obtained from diffusion calculations, under the non-interacting ion approximation, and with experiment. This method is shown to provide good quantitative estimates for the ionic mobilities of two silver conductors, [Formula: see text]-AgI and [Formula: see text]-RbAg[Formula: see text]I[Formula: see text]. In addition to being convenient and numerically robust, this method accounts for ion-ion correlations at a much lower computational cost than exact approaches.
我们描述了一种基于非平衡分子动力学(NEMD)模拟的方法,用于从第一性原理计算固体离子导体(如固体电解质)的离子迁移率。计算在材料的有限平板中进行,在平板中施加电场并测量移动离子的动态响应。我们将我们的结果与在非相互作用离子近似下从扩散计算得到的结果以及实验结果进行比较。结果表明,该方法能为两种银导体α-AgI和β-RbAg₄I₅的离子迁移率提供良好的定量估计。除了方便且数值稳健外,该方法以比精确方法低得多的计算成本考虑了离子-离子相关性。