Sanko University, School of Medicine, Gaziantep, Turkey.
Department of Pediatrics, University of Kentucky College of Medicine, Lexington, KY, USA; Department of Pharmacology and Nutritional Sciences, University of Kentucky, Lexington, KY, USA.
J Nutr Biochem. 2023 Apr;114:109224. doi: 10.1016/j.jnutbio.2022.109224. Epub 2022 Nov 18.
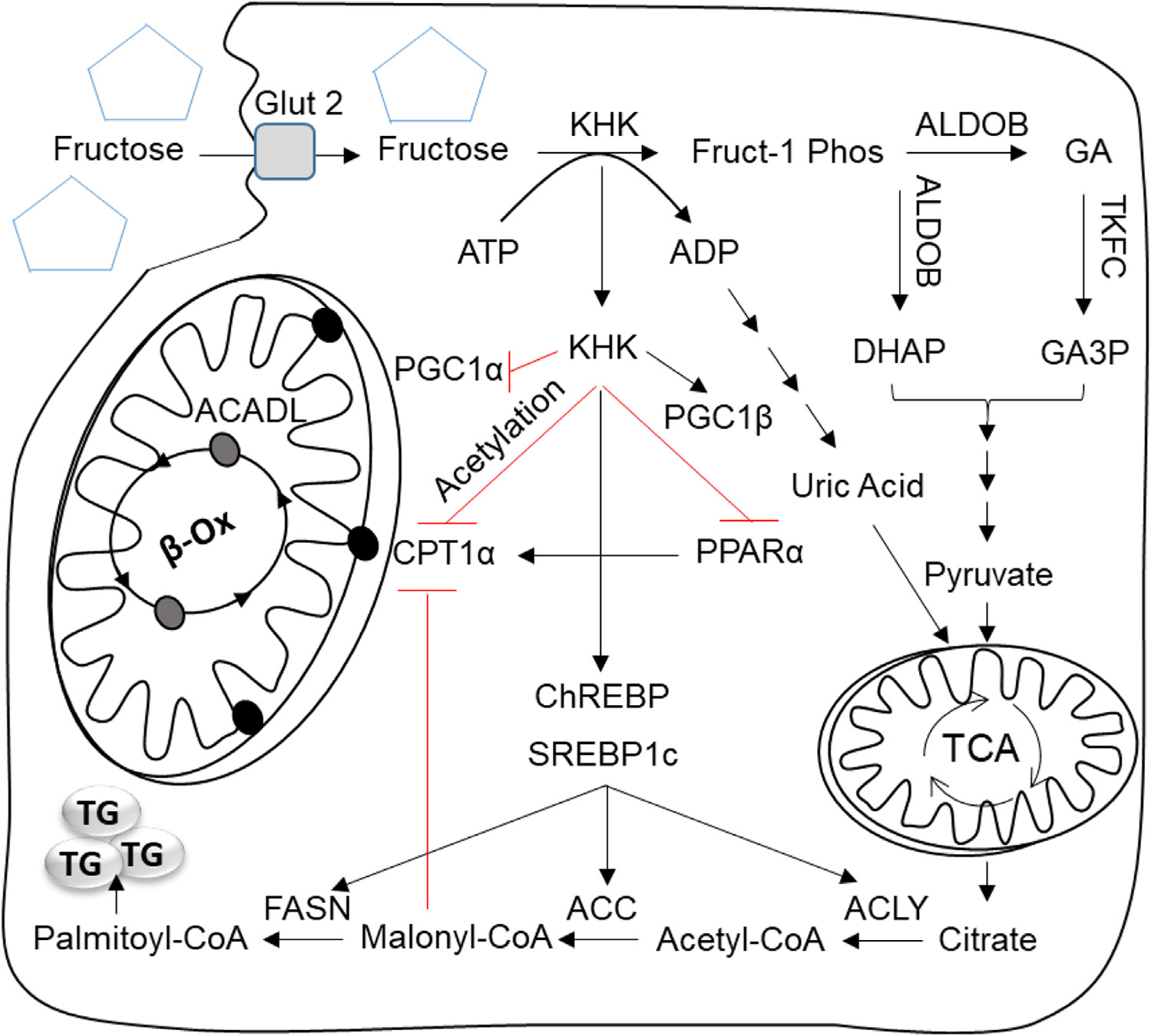
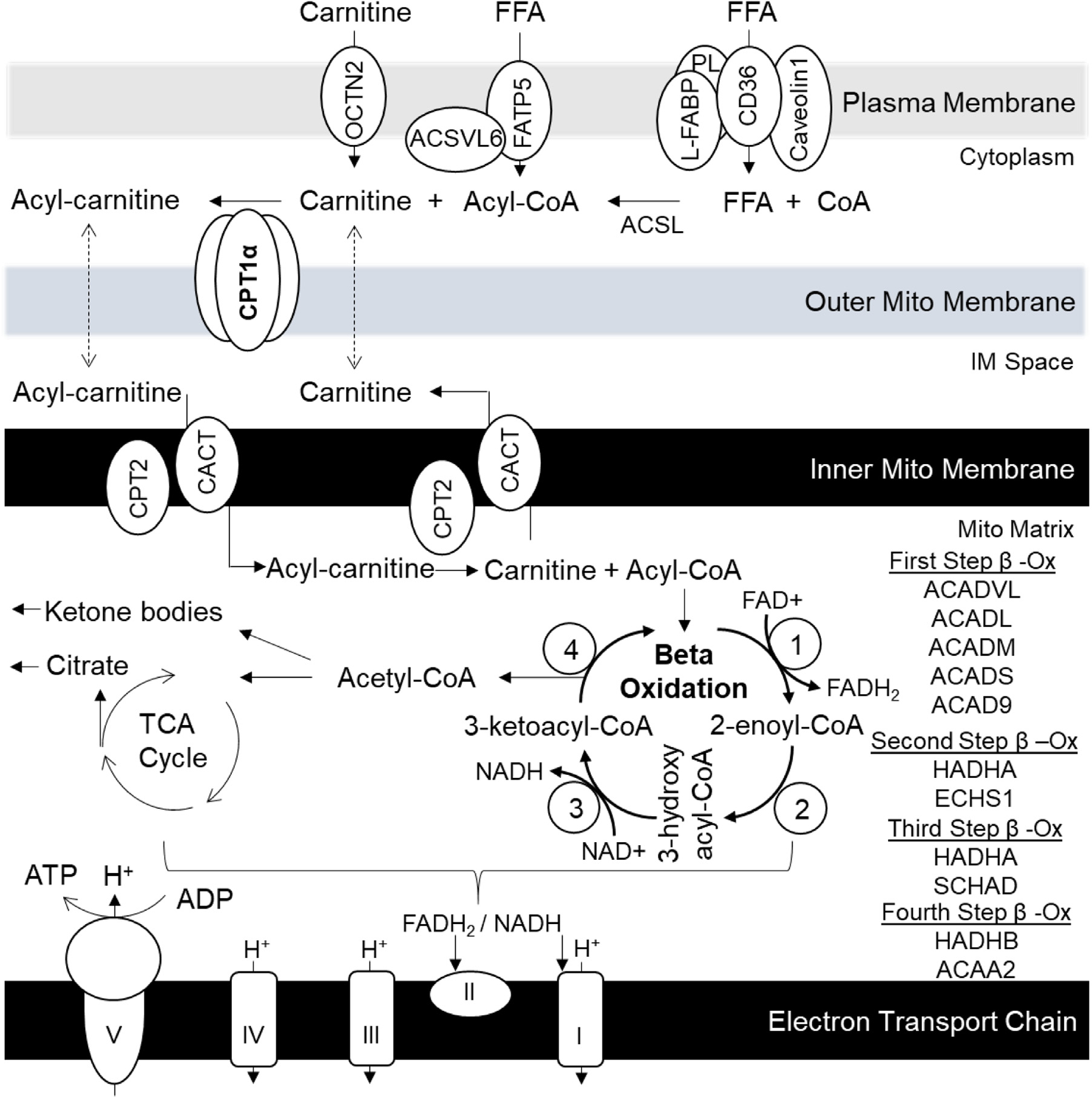
Increased fructose intake from sugar-sweetened beverages and highly processed sweets is a well-recognized risk factor for the development of obesity and its complications. Fructose strongly supports lipogenesis on a normal chow diet by providing both, a substrate for lipid synthesis and activation of lipogenic transcription factors. However, the negative health consequences of dietary sugar are best observed with the concomitant intake of a HFD. Indeed, the most commonly used obesogenic research diets, such as "Western diet", contain both fructose and a high amount of fat. In spite of its common use, how the combined intake of fructose and fat synergistically supports development of metabolic complications is not fully elucidated. Here we present the preponderance of evidence that fructose consumption decreases oxidation of dietary fat in human and animal studies. We provide a detailed review of the mitochondrial β-oxidation pathway. Fructose affects hepatic activation of fatty acyl-CoAs, decreases acylcarnitine production and impairs the carnitine shuttle. Mechanistically, fructose suppresses transcriptional activity of PPARα and its target CPT1α, the rate limiting enzyme of acylcarnitine production. These effects of fructose may be, in part, mediated by protein acetylation. Acetylation of PGC1α, a co-activator of PPARα and acetylation of CPT1α, in part, account for fructose-impaired acylcarnitine production. Interestingly, metabolic effects of fructose in the liver can be largely overcome by carnitine supplementation. In summary, fructose decreases oxidation of dietary fat in the liver, in part, by impairing acylcarnitine production, offering one explanation for the synergistic effects of these nutrients on the development of metabolic complications, such as NAFLD.
增加糖饮料和高度加工甜食中的果糖摄入是肥胖及其并发症发展的公认危险因素。果糖通过提供脂质合成的底物和激活脂肪生成转录因子,强烈支持正常饲料中的脂肪生成。然而,饮食糖的负面健康后果在同时摄入高脂肪饮食时观察得最为明显。事实上,最常用的肥胖研究饮食,如“西方饮食”,既含有果糖又含有大量脂肪。尽管果糖被广泛使用,但果糖和脂肪的联合摄入如何协同支持代谢并发症的发展尚不完全清楚。在这里,我们提出了大量的证据,表明果糖的摄入会降低人类和动物研究中膳食脂肪的氧化。我们详细回顾了线粒体β-氧化途径。果糖影响肝脂肪酸酰基辅酶 A 的激活,减少酰基肉碱的产生,并损害肉碱穿梭系统。从机制上讲,果糖抑制了 PPARα及其靶标 CPT1α的转录活性,CPT1α是酰基肉碱生成的限速酶。果糖的这些作用部分可能是通过蛋白乙酰化介导的。果糖抑制了 PPARα的共激活因子 PGC1α的转录活性,以及 CPT1α的乙酰化,部分解释了果糖对酰基肉碱生成的损害作用。有趣的是,肝脏中果糖的代谢作用可以通过肉碱补充来很大程度上克服。总之,果糖通过损害酰基肉碱的生成,部分降低了肝脏中膳食脂肪的氧化,为这些营养素在代谢并发症(如非酒精性脂肪性肝病)发展中的协同作用提供了一种解释。