Genetics and Genome Biology Program, Research Institute, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada.
Division of Clinical and Metabolic Genetics, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada.
Hum Mol Genet. 2023 Apr 20;32(9):1429-1438. doi: 10.1093/hmg/ddac289.
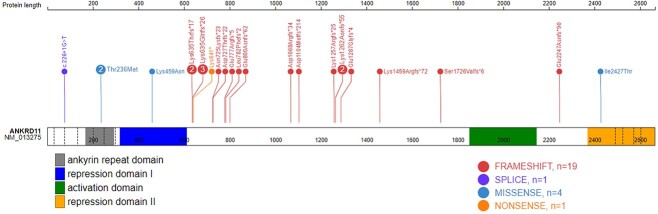
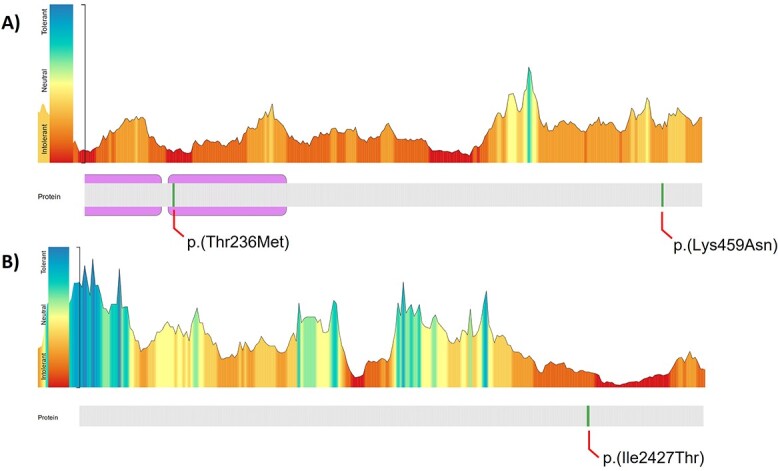
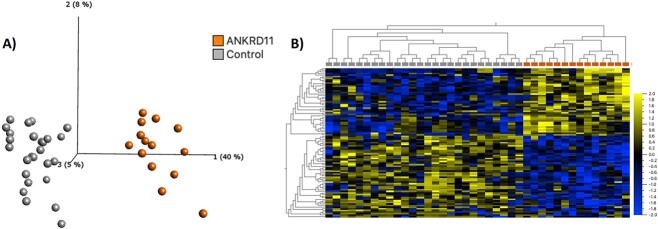
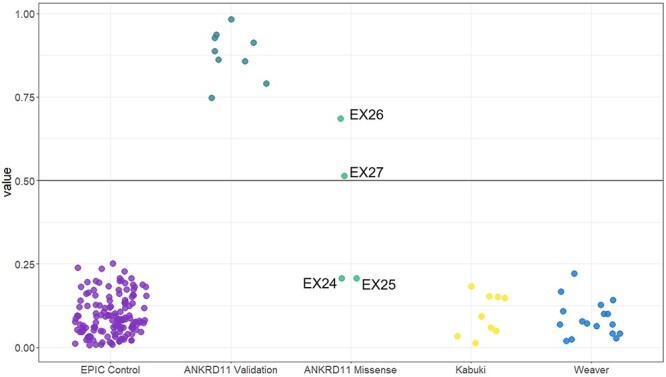
Pathogenic variants in ANKRD11 or microdeletions at 16q24.3 are the cause of KBG syndrome (KBGS), a neurodevelopmental syndrome characterized by intellectual disability, dental and skeletal anomalies, and characteristic facies. The ANKRD11 gene encodes the ankyrin repeat-containing protein 11A transcriptional regulator, which is expressed in the brain and implicated in neural development. Syndromic conditions caused by pathogenic variants in epigenetic regulatory genes show unique patterns of DNA methylation (DNAm) in peripheral blood, termed DNAm signatures. Given ANKRD11's role in chromatin modification, we tested whether pathogenic ANKRD11 variants underlying KBGS are associated with a DNAm signature. We profiled whole-blood DNAm in 21 individuals with ANKRD11 variants, 2 individuals with microdeletions at 16q24.3 and 28 typically developing individuals, using Illumina's Infinium EPIC array. We identified 95 differentially methylated CpG sites that distinguished individuals with KBGS and pathogenic variants in ANKRD11 (n = 14) from typically developing controls (n = 28). This DNAm signature was then validated in an independent cohort of seven individuals with KBGS and pathogenic ANKRD11 variants. We generated a machine learning model from the KBGS DNAm signature and classified the DNAm profiles of four individuals with variants of uncertain significance (VUS) in ANKRD11. We identified an intermediate classification score for an inherited missense variant transmitted from a clinically unaffected mother to her affected child. In conclusion, we show that the DNAm profiles of two individuals with 16q24.3 microdeletions were indistinguishable from the DNAm profiles of individuals with pathogenic variants in ANKRD11, and we demonstrate the diagnostic utility of the new KBGS signature by classifying the DNAm profiles of individuals with VUS in ANKRD11.
ANKRD11 中的致病变异或 16q24.3 处的微缺失是 KBG 综合征(KBGS)的病因,这是一种神经发育综合征,其特征为智力残疾、牙齿和骨骼异常以及特征性面容。ANKRD11 基因编码含锚蛋白重复结构域的蛋白 11A 转录调节因子,该蛋白在大脑中表达,与神经发育有关。由表观遗传调控基因中的致病性变异引起的综合征表现出外周血中独特的 DNA 甲基化(DNAm)模式,称为 DNAm 特征。鉴于 ANKRD11 在染色质修饰中的作用,我们测试了导致 KBGS 的致病性 ANKRD11 变异是否与 DNAm 特征相关。我们使用 Illumina 的 Infinium EPIC 阵列对 21 名携带 ANKRD11 变异的个体、2 名携带 16q24.3 微缺失的个体和 28 名典型发育个体的全血 DNAm 进行了分析。我们确定了 95 个差异甲基化 CpG 位点,这些位点可将 KBGS 患者和携带致病性 ANKRD11 变异的个体(n=14)与典型发育对照组(n=28)区分开来。该 DNAm 特征随后在 7 名携带 KBGS 和致病性 ANKRD11 变异的个体的独立队列中进行了验证。我们从 KBGS DNAm 特征中生成了一个机器学习模型,并对携带 ANKRD11 变异的意义不明的 4 名个体的 DNAm 谱进行了分类。我们确定了一种从中继的错义变体继承的中间分类评分,该变体从临床无影响的母亲传递给受影响的孩子。总之,我们表明,2 名携带 16q24.3 微缺失的个体的 DNAm 谱与携带 ANKRD11 致病性变异的个体的 DNAm 谱无法区分,我们通过对携带 ANKRD11 变异的意义不明的个体的 DNAm 谱进行分类,证明了新的 KBGS 特征的诊断效用。