Roth Daniela Marta, Baddam Pranidhi, Lin Haiming, Vidal-García Marta, Aponte Jose David, De Souza Sarah-Thea, Godziuk Devyn, Watson Adrianne Eve Scovil, Footz Tim, Schachter Nathan F, Egan Sean E, Hallgrímsson Benedikt, Graf Daniel, Voronova Anastassia
School of Dentistry, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada.
Department of Cell Biology & Anatomy, Alberta Children's Hospital Research Institute, University of Calgary, Calgary, AB, Canada.
Front Cell Dev Biol. 2021 Apr 29;9:645386. doi: 10.3389/fcell.2021.645386. eCollection 2021.
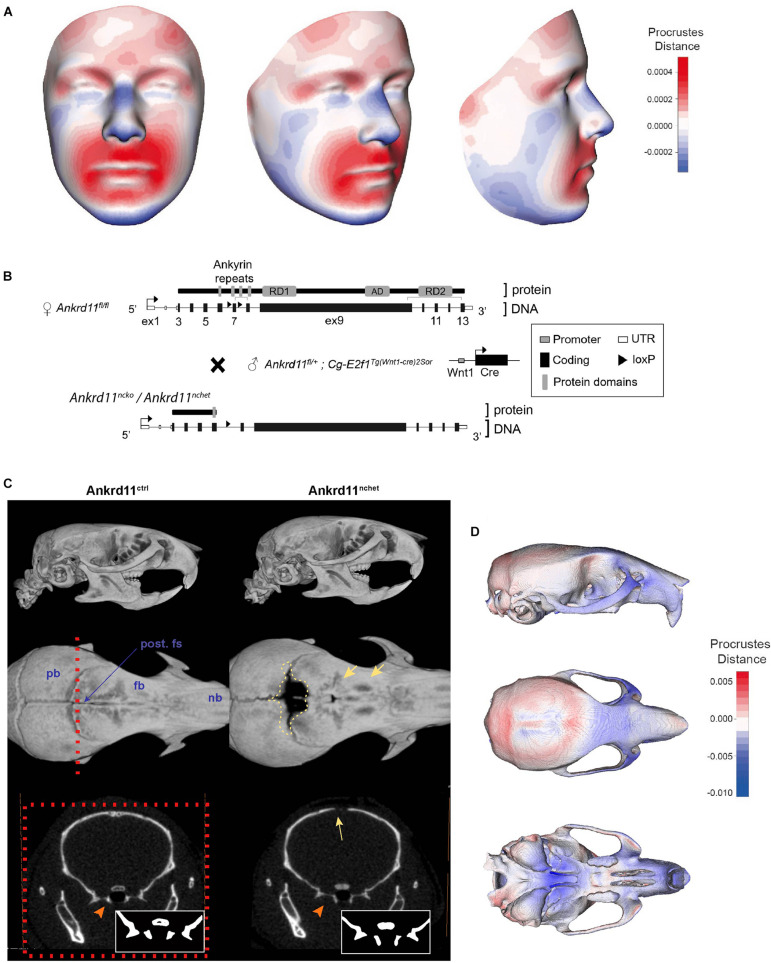
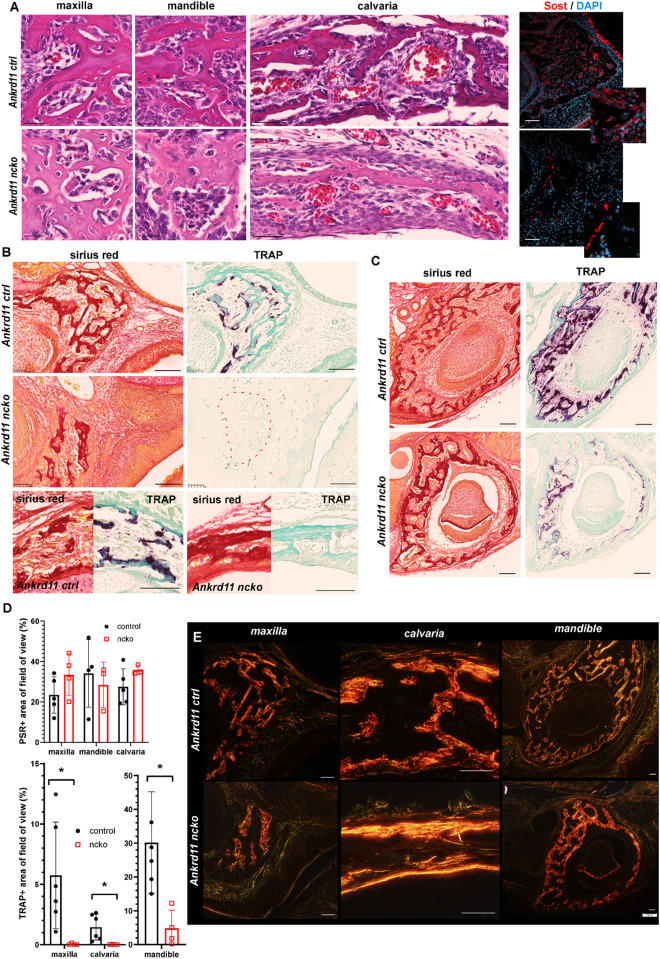
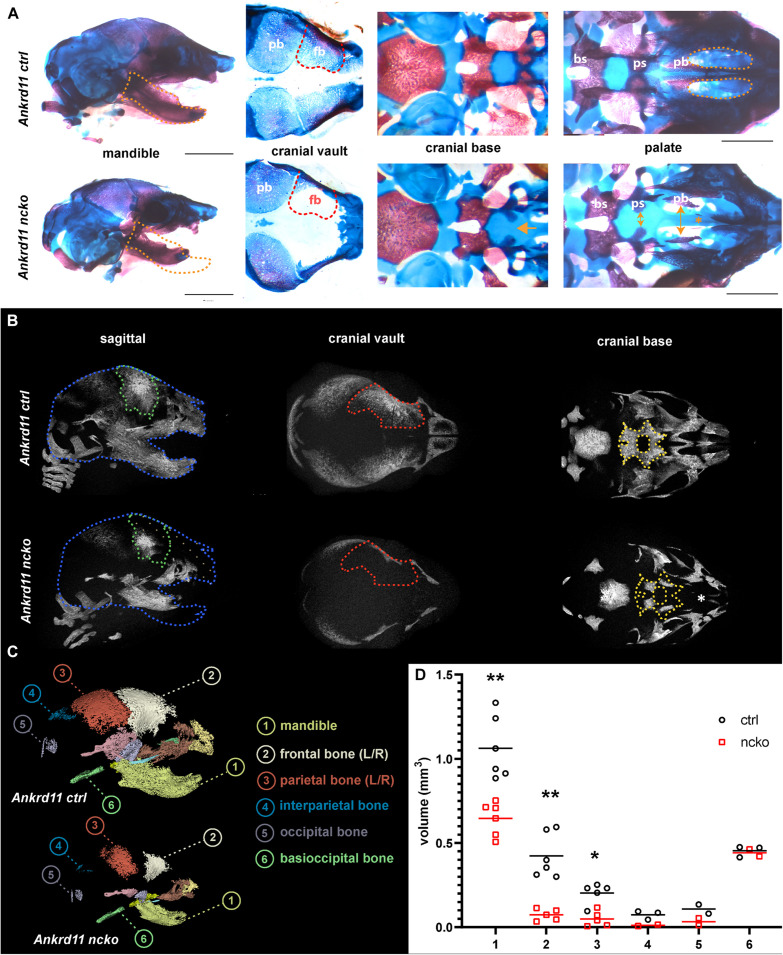
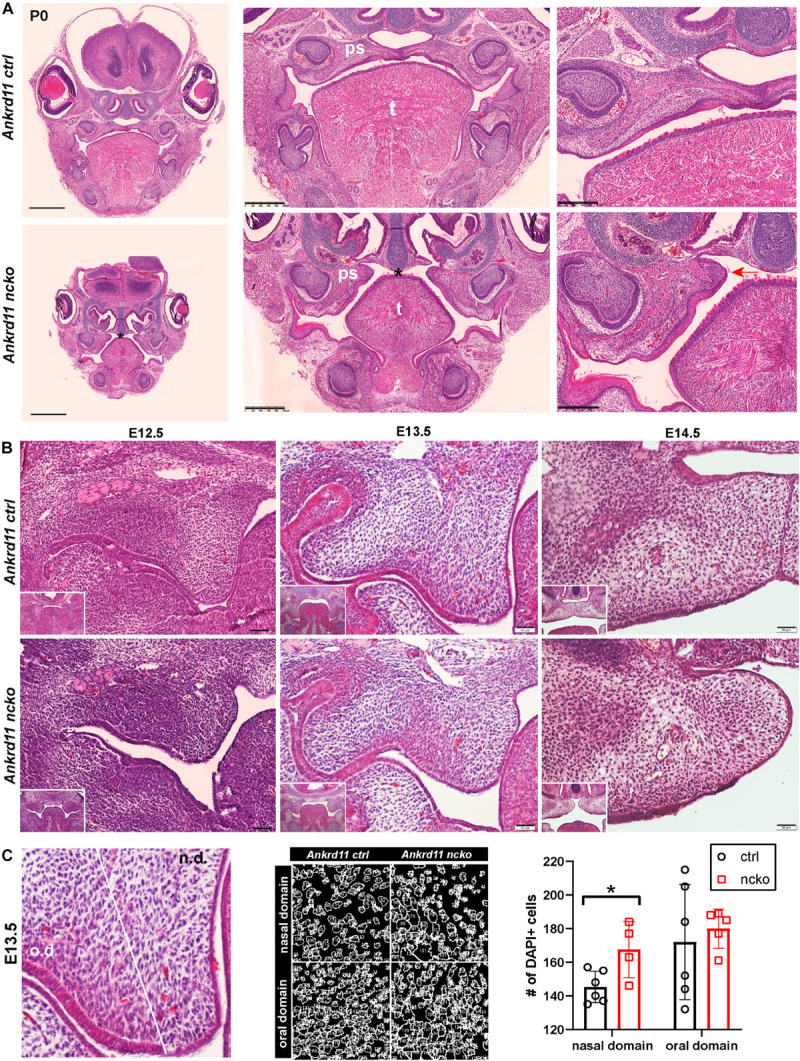
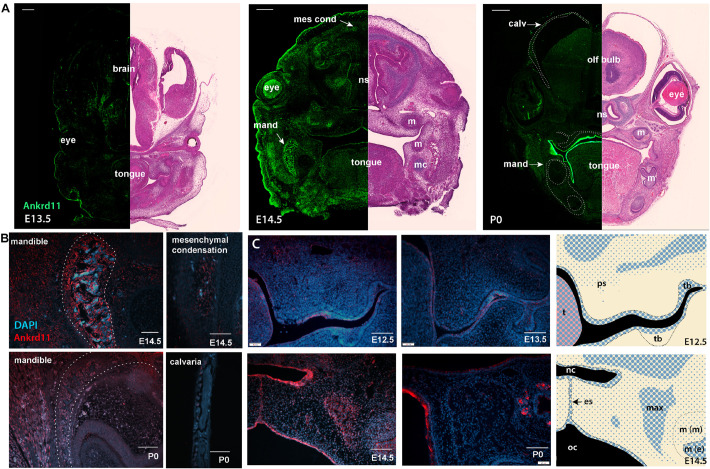
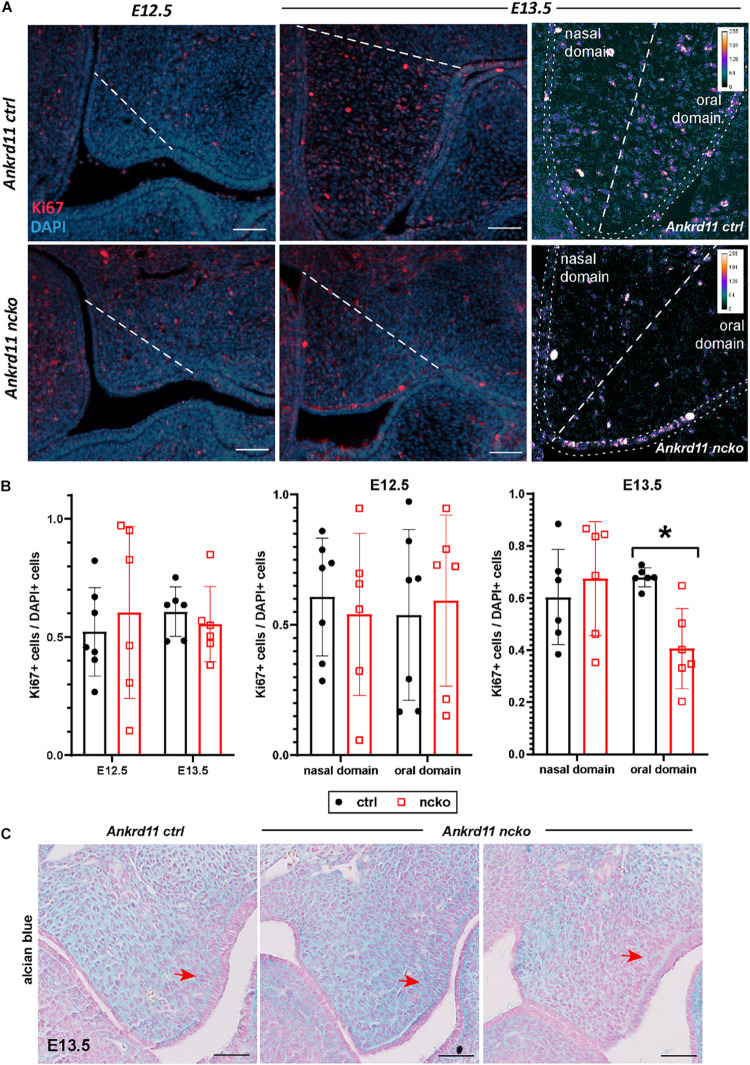
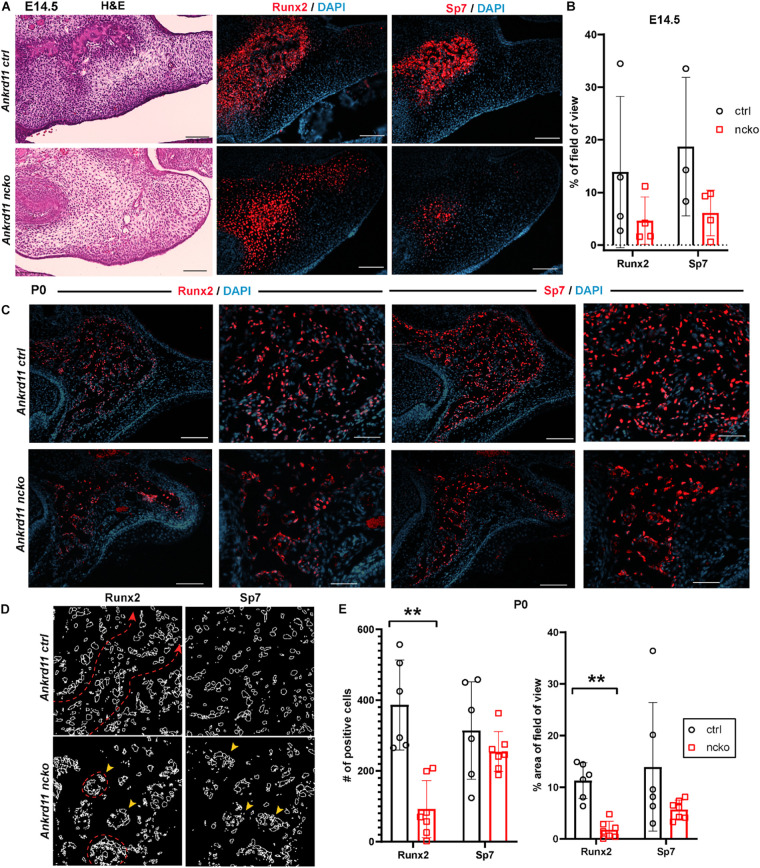
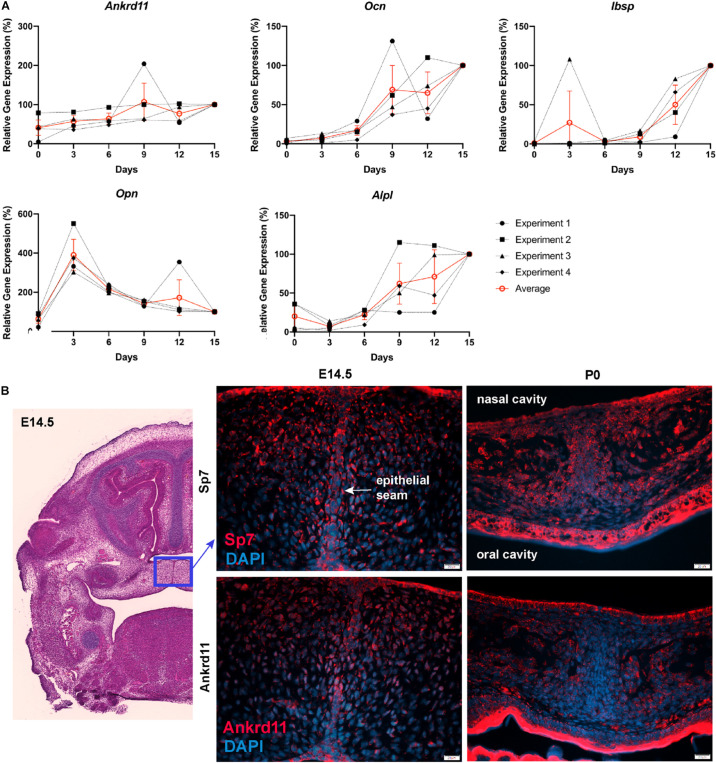
Epigenetic and chromatin regulation of craniofacial development remains poorly understood. Ankyrin Repeat Domain 11 () is a chromatin regulator that has previously been shown to control neural stem cell fates via modulation of histone acetylation. gene variants, or microdeletions of the 16q24.3 chromosomal region encompassing the gene, cause KBG syndrome, a rare autosomal dominant congenital disorder with variable neurodevelopmental and craniofacial involvement. Craniofacial abnormalities include a distinct facial gestalt, delayed bone age, tooth abnormalities, delayed fontanelle closure, and frequently cleft or submucosal palate. Despite this, the dramatic phenotype and precise role of in embryonic craniofacial development remain unexplored. Quantitative analysis of 3D images of KBG syndromic subjects shows an overall reduction in the size of the middle and lower face. Here, we report that mice with heterozygous deletion of in neural crest cells (Ankrd11) display a mild midfacial hypoplasia including reduced midfacial width and a persistent open fontanelle, both of which mirror KBG syndrome patient facial phenotypes. Mice with a homozygous deletion in neural crest cells (Ankrd11) die at birth. They show increased severity of several clinical manifestations described for KBG syndrome, such as cleft palate, retrognathia, midfacial hypoplasia, and reduced calvarial growth. At E14.5, expression in the craniofacial complex is closely associated with developing bony structures, while expression at birth is markedly decreased. Conditional deletion of leads to a reduction in ossification of midfacial bones, with several ossification centers failing to expand and/or fuse. Intramembranous bones show features of delayed maturation, with bone remodeling severely curtailed at birth. Palatal shelves remain hypoplastic at all developmental stages, with a local reduction in proliferation at E13.5. Our study identifies as a critical regulator of intramembranous ossification and palate development and suggests that Ankrd11 and Ankrd11 mice may serve as pre-clinical models for KBG syndrome in humans.
颅面发育的表观遗传和染色质调控仍未得到充分理解。锚蛋白重复结构域11(ANKRD11)是一种染色质调节因子,先前已被证明可通过调节组蛋白乙酰化来控制神经干细胞的命运。ANKRD11基因变异或包含ANKRD11基因的16q24.3染色体区域的微缺失会导致KBG综合征,这是一种罕见的常染色体显性先天性疾病,伴有可变的神经发育和颅面受累。颅面异常包括独特的面部形态、骨龄延迟、牙齿异常、囟门闭合延迟,以及频繁的腭裂或黏膜下腭裂。尽管如此,ANKRD11在胚胎颅面发育中的显著表型和精确作用仍未被探索。对KBG综合征患者三维图像的定量分析显示,中下面部尺寸总体减小。在此,我们报告神经嵴细胞中ANKRD11杂合缺失的小鼠(Ankrd11+/-)表现出轻度的面中部发育不全,包括面中部宽度减小和囟门持续开放,这两者都与KBG综合征患者的面部表型相似。神经嵴细胞中ANKRD11纯合缺失的小鼠(Ankrd11-/-)在出生时死亡。它们表现出KBG综合征所描述的几种临床表现的严重程度增加,如腭裂、下颌后缩、面中部发育不全和颅骨生长减少。在胚胎第14.5天,ANKRD11在颅面复合体中的表达与正在发育的骨结构密切相关,而出生时的表达则明显降低。ANKRD11的条件性缺失导致面中部骨骼骨化减少,几个骨化中心未能扩展和/或融合。膜内骨显示出成熟延迟的特征,出生时骨重塑严重减少。腭突在所有发育阶段都发育不全,在胚胎第13.5天局部增殖减少。我们的研究确定ANKRD11是膜内骨化和腭发育的关键调节因子,并表明Ankrd11+/-和Ankrd11-/-小鼠可能作为人类KBG综合征的临床前模型。