Molecular Genetics Thalassaemia Department, The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus.
Leiden University Medical Center, Leiden, Netherlands.
Elife. 2022 Dec 1;11:e79713. doi: 10.7554/eLife.79713.
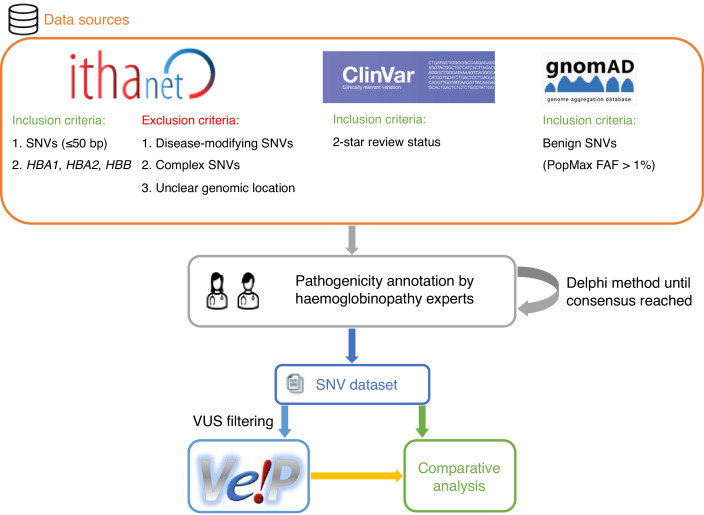
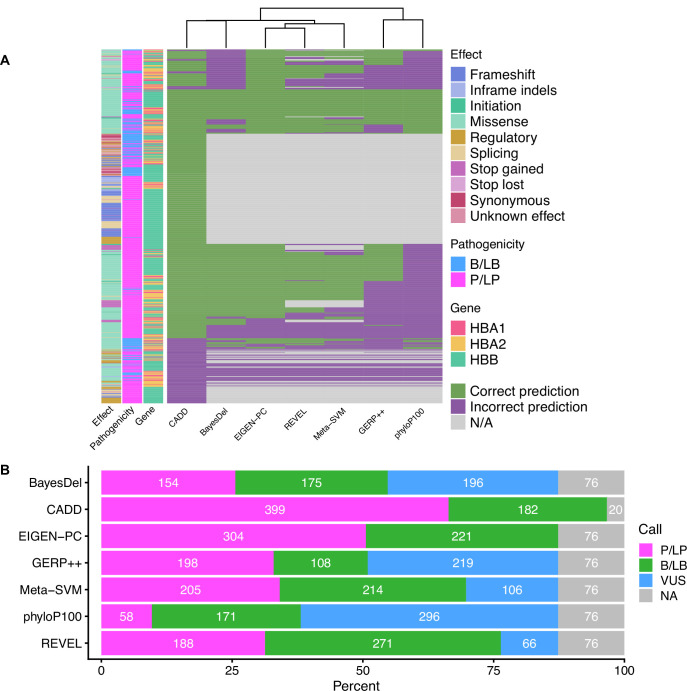
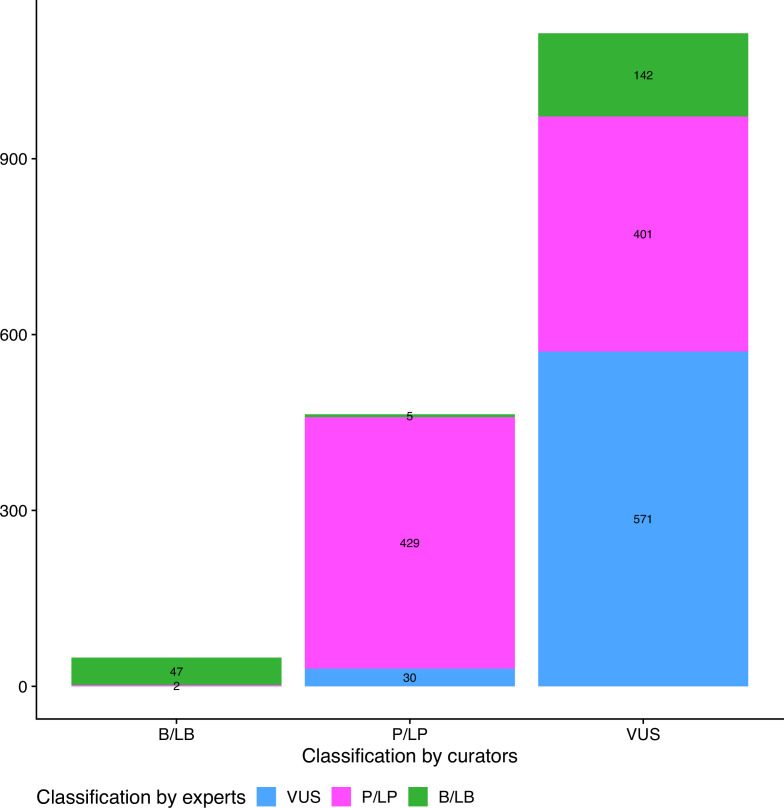
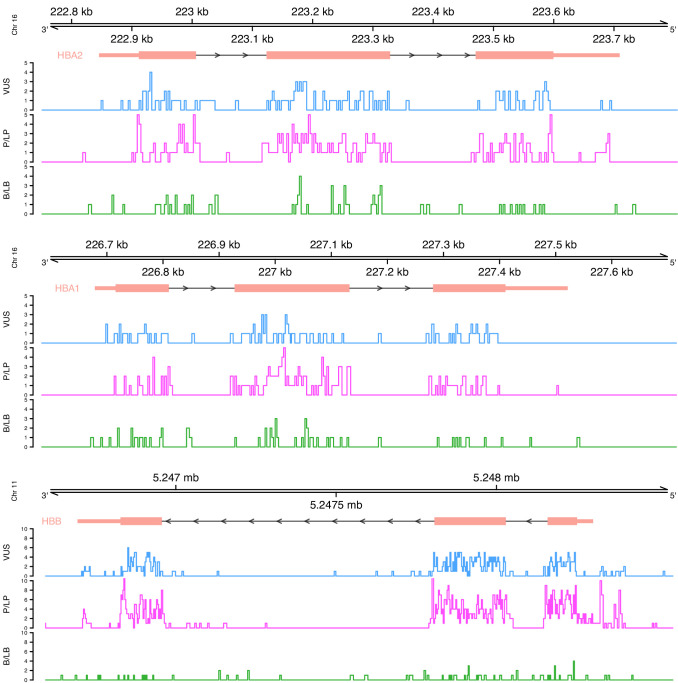
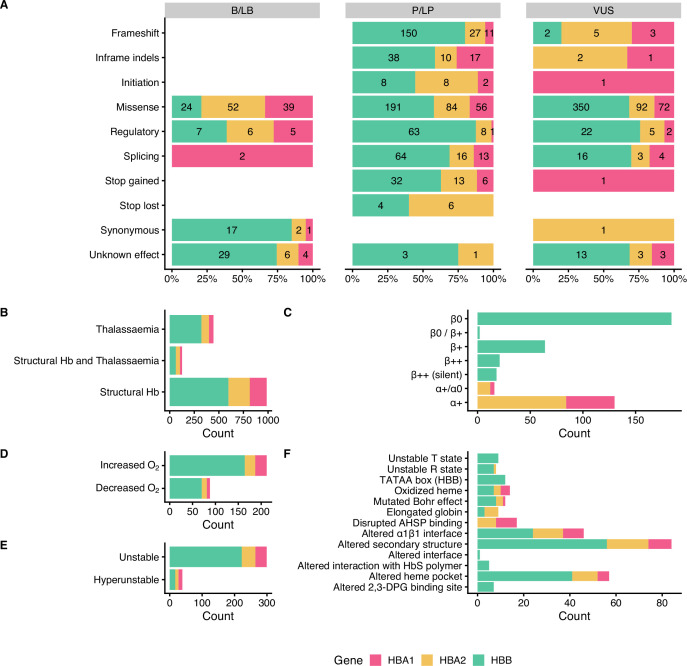
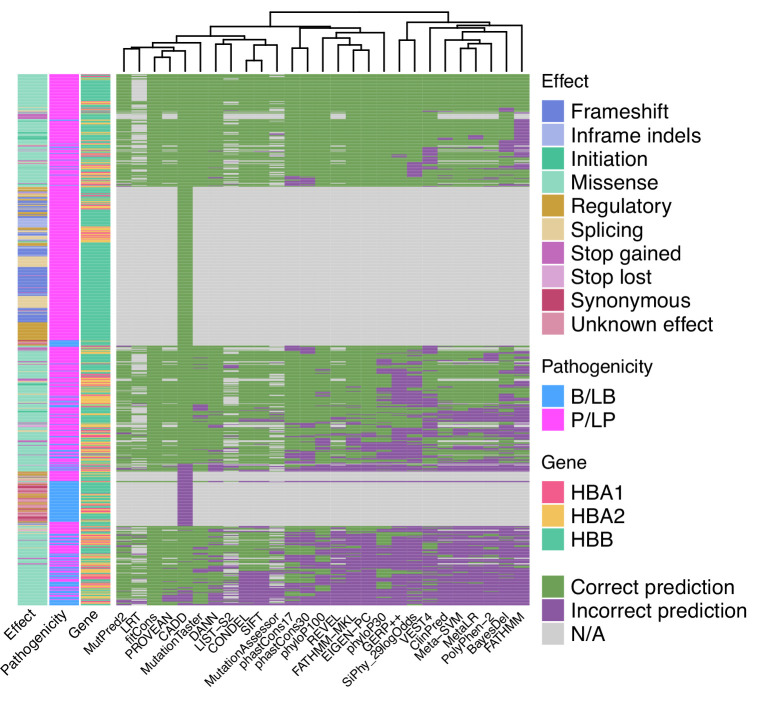
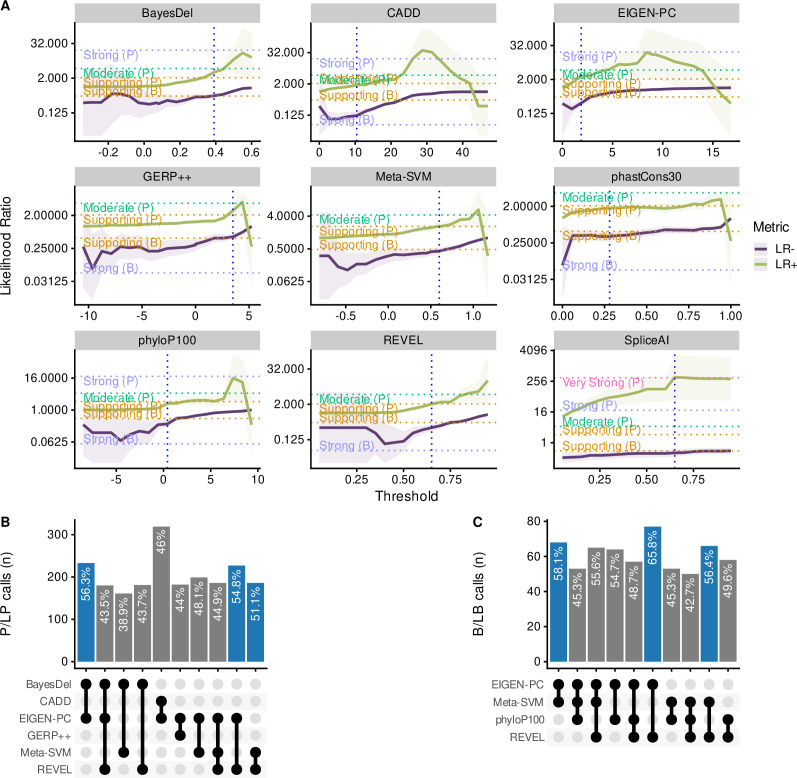
Haemoglobinopathies are the commonest monogenic diseases worldwide and are caused by variants in the globin gene clusters. With over 2400 variants detected to date, their interpretation using the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines is challenging and computational evidence can provide valuable input about their functional annotation. While many in silico predictors have already been developed, their performance varies for different genes and diseases. In this study, we evaluate 31 in silico predictors using a dataset of 1627 variants in , and . By varying the decision threshold for each tool, we analyse their performance (a) as binary classifiers of pathogenicity and (b) by using different non-overlapping pathogenic and benign thresholds for their optimal use in the ACMG/AMP framework. Our results show that CADD, Eigen-PC, and REVEL are the overall top performers, with the former reaching moderate strength level for pathogenic prediction. Eigen-PC and REVEL achieve the highest accuracies for missense variants, while CADD is also a reliable predictor of non-missense variants. Moreover, SpliceAI is the top performing splicing predictor, reaching strong level of evidence, while GERP++ and phyloP are the most accurate conservation tools. This study provides evidence about the optimal use of computational tools in globin gene clusters under the ACMG/AMP framework.
血红蛋白病是全球最常见的单基因疾病,由珠蛋白基因簇中的变异引起。迄今为止,已经检测到超过 2400 种变异,使用美国医学遗传学与基因组学学院(ACMG)/分子病理学协会(AMP)指南进行解释具有挑战性,而计算证据可以为其功能注释提供有价值的信息。虽然已经开发了许多计算预测工具,但它们在不同基因和疾病中的性能存在差异。在这项研究中,我们使用了 1627 种 、 和 变体的数据集,评估了 31 种计算预测工具。通过为每个工具改变决策阈值,我们分析了它们的性能:(a) 作为致病性的二进制分类器,(b) 通过为 ACMG/AMP 框架的最佳使用使用不同的非重叠致病性和良性阈值。我们的研究结果表明,CADD、Eigen-PC 和 REVEL 是整体表现最佳的工具,前者在致病性预测方面达到了中等强度水平。Eigen-PC 和 REVEL 对错义变异具有最高的准确性,而 CADD 也是非错义变异的可靠预测工具。此外,SpliceAI 是表现最佳的剪接预测工具,达到了强有力的证据水平,而 GERP++和 phyloP 是最准确的保守性工具。本研究为在 ACMG/AMP 框架下在珠蛋白基因簇中最佳使用计算工具提供了证据。