Genomic Informatics Group, Human Development and Health, Faculty of Medicine, University Hospital Southampton, MP 808, Duthie Building, Southampton, SO16 6YD, Hampshire, UK.
Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, 02142, USA.
Hum Genet. 2023 Mar;142(3):351-362. doi: 10.1007/s00439-022-02509-x. Epub 2022 Dec 7.
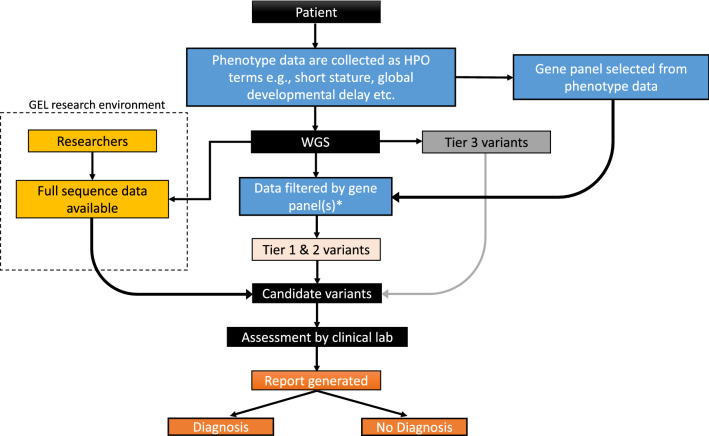
Genome sequencing was first offered clinically in the UK through the 100,000 Genomes Project (100KGP). Analysis was restricted to predefined gene panels associated with the patient's phenotype. However, panels rely on clearly characterised phenotypes and risk missing diagnoses outside of the panel(s) applied. We propose a complementary method to rapidly identify pathogenic variants, including those missed by 100KGP methods.
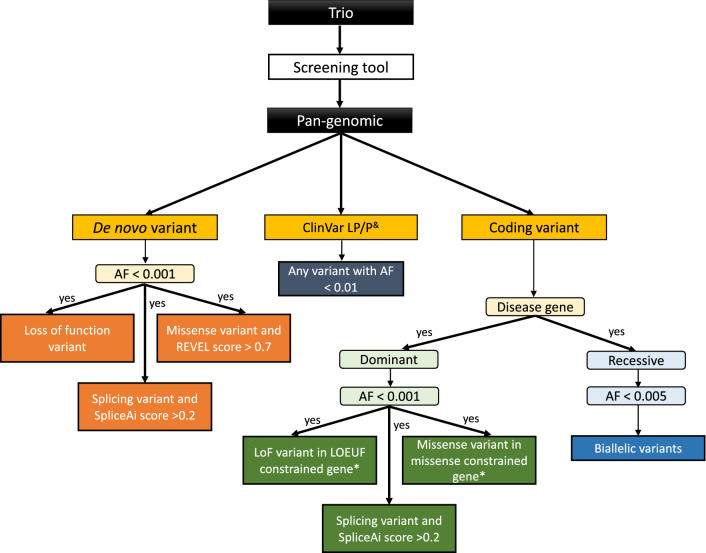
The Loss-of-function Observed/Expected Upper-bound Fraction (LOEUF) score quantifies gene constraint, with low scores correlated with haploinsufficiency. We applied DeNovoLOEUF, a filtering strategy to sequencing data from 13,949 rare disease trios in the 100KGP, by filtering for rare, de novo, loss-of-function variants in disease genes with a LOEUF score < 0.2. We compared our findings with the corresponding patient's diagnostic reports.
324/332 (98%) of the variants identified using DeNovoLOEUF were diagnostic or partially diagnostic (whereby the variant was responsible for some of the phenotype). We identified 39 diagnoses that were "missed" by 100KGP standard analyses, which are now being returned to patients.
We have demonstrated a highly specific and rapid method with a 98% positive predictive value that has good concordance with standard analysis, low false-positive rate, and can identify additional diagnoses. Globally, as more patients are being offered genome sequencing, we anticipate that DeNovoLOEUF will rapidly identify new diagnoses and facilitate iterative analyses when new disease genes are discovered.
基因组测序最初通过英国的 10 万基因组计划(100KGP)在临床上提供。分析仅限于与患者表型相关的预定义基因面板。然而,面板依赖于明确表征的表型,并且可能会错过应用面板之外的诊断。我们提出了一种补充方法,可以快速识别致病性变体,包括 100KGP 方法错过的变体。
失活观察/预期上限分数(LOEUF)评分量化基因约束,低分数与单倍不足相关。我们应用了 DeNovoLOEUF,这是一种过滤策略,用于对 100KGP 中 13949 个罕见疾病三亲体的测序数据进行过滤,方法是对 LOEUF 评分<0.2 的疾病基因中的稀有、新生、失活变体进行过滤。我们将我们的发现与相应患者的诊断报告进行了比较。
使用 DeNovoLOEUF 鉴定的 324/332(98%)变体是诊断性或部分诊断性的(即变体导致部分表型)。我们确定了 39 个 100KGP 标准分析“遗漏”的诊断,现在将这些诊断返回给患者。
我们已经证明了一种具有 98%阳性预测值的高度特异性和快速方法,与标准分析具有良好的一致性,假阳性率低,并可以识别其他诊断。在全球范围内,随着越来越多的患者接受基因组测序,我们预计 DeNovoLOEUF 将迅速识别新的诊断,并在发现新的疾病基因时促进迭代分析。