Seaby Eleanor G, Leggatt Gary, Cheng Guo, Thomas N Simon, Ashton James J, Stafford Imogen, Baralle Diana, Rehm Heidi L, O'Donnell-Luria Anne, Ennis Sarah
Human Development and Health, Faculty of Medicine, University Hospital Southampton, Southampton, Hampshire, SO16 6YD, UK.
Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA.
medRxiv. 2023 Mar 30:2023.03.21.23287545. doi: 10.1101/2023.03.21.23287545.
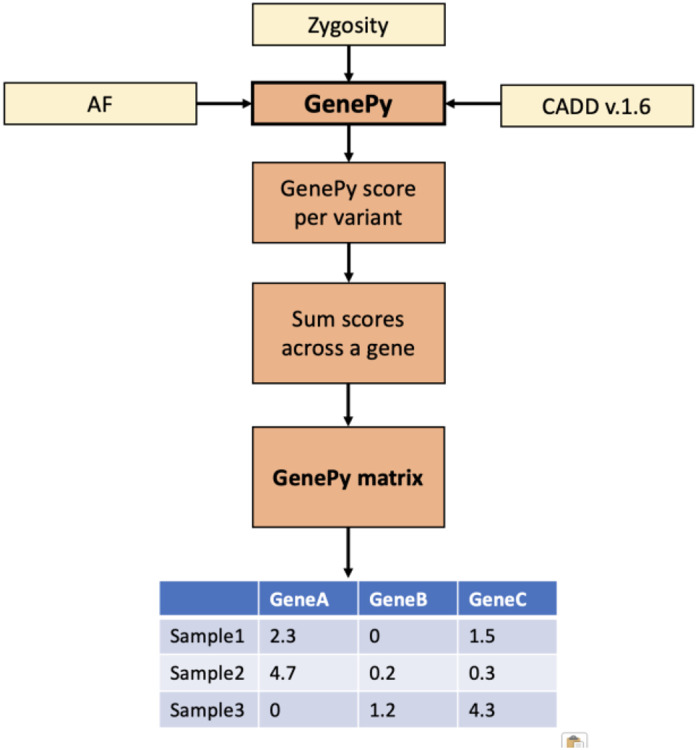
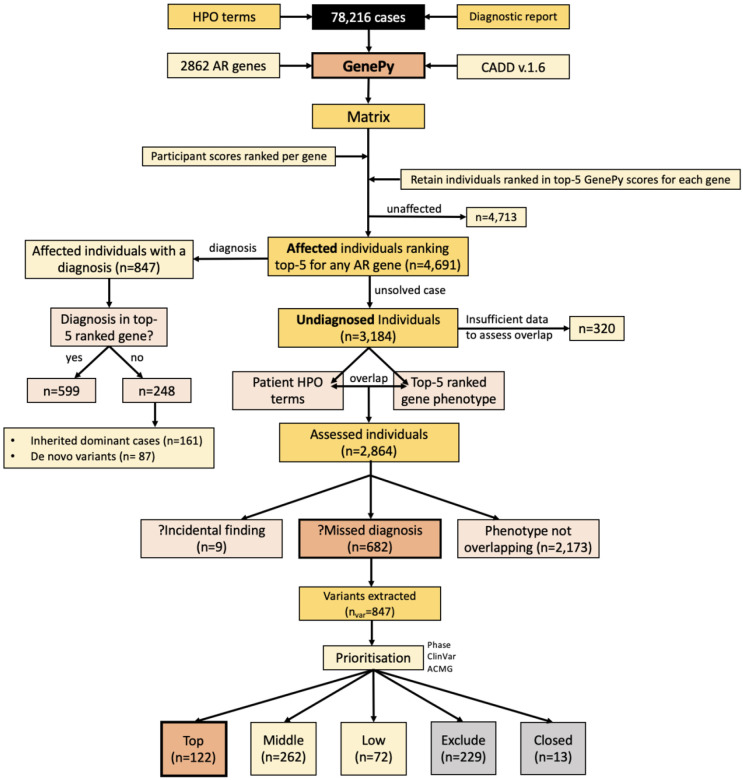
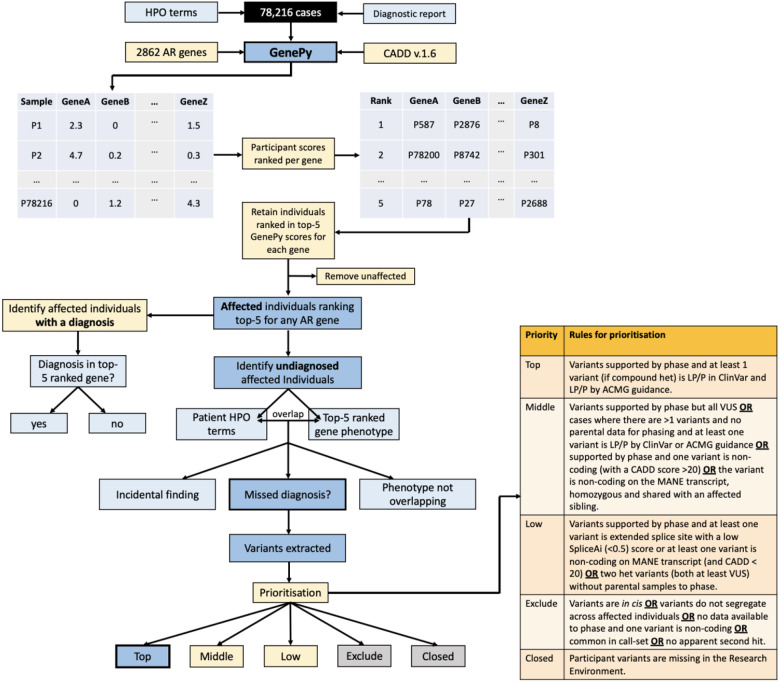
The 100,000 Genomes Project (100KGP) diagnosed a quarter of recruited affected participants, but 26% of diagnoses were in genes not on the chosen gene panel(s); with many being variants of high impact. However, assessing biallelic variants without a gene panel is challenging, due to the number of variants requiring scrutiny. We sought to identify potential missed biallelic diagnoses independent of the gene panel applied using GenePy - a whole gene pathogenicity metric. GenePy scores all variants called in a given individual, incorporating allele frequency, zygosity, and a user-defined deleterious metric (CADD v1.6 applied herein). GenePy then combines all variant scores for individual genes, generating an aggregate score per gene, per participant. We calculated GenePy scores for 2862 recessive disease genes in 78,216 individuals in 100KGP. For each gene, we ranked participant GenePy scores for that gene, and scrutinised affected individuals without a diagnosis whose scores ranked amongst the top-5 for each gene. We assessed these participants' phenotypes for overlap with the disease gene associated phenotype for which they were highly ranked. Where phenotypes overlapped, we extracted rare variants in the gene of interest and applied phase, ClinVar and ACMG classification looking for putative causal biallelic variants. 3184 affected individuals without a molecular diagnosis had a top-5 ranked GenePy gene score and 682/3184 (21%) had phenotypes overlapping with one of the top-ranking genes. After removing 13 withdrawn participants, in 122/669 (18%) of the phenotype-matched cases, we identified a putative missed diagnosis in a top-ranked gene supported by phasing, ClinVar and ACMG classification. A further 334/669 (50%) of cases have a possible missed diagnosis but require functional validation. Applying GenePy at scale has identified potential diagnoses for 456/3183 (14%) of undiagnosed participants who had a top-5 ranked GenePy score in a recessive disease gene, whilst adding only 1.2 additional variants (per individual) for assessment.
“十万人基因组计划”(100KGP)诊断出了四分之一的招募到的患病参与者,但26%的诊断结果是在所选基因panel之外的基因中;其中许多是具有高影响的变异。然而,由于需要仔细审查的变异数量众多,在没有基因panel的情况下评估双等位基因变异具有挑战性。我们试图使用GenePy(一种全基因致病性指标)来识别与所应用的基因panel无关的潜在遗漏双等位基因诊断。GenePy对给定个体中检测到的所有变异进行评分,纳入等位基因频率、纯合度和用户定义的有害性指标(本文应用CADD v1.6)。然后,GenePy将单个基因的所有变异分数进行合并,为每个基因、每个参与者生成一个综合分数。我们计算了100KGP中78216名个体中2862个隐性疾病基因的GenePy分数。对于每个基因,我们对该基因的参与者GenePy分数进行排名,并仔细审查那些在每个基因中分数排名前5但未得到诊断的患病个体。我们评估了这些参与者的表型与他们排名靠前的疾病基因相关表型的重叠情况。当表型重叠时,我们提取感兴趣基因中的罕见变异,并应用相位分析、ClinVar和ACMG分类来寻找假定的致病双等位基因变异。3184名未得到分子诊断的患病个体具有排名前5的GenePy基因分数,其中682/3184(21%)的表型与排名靠前的基因之一重叠。在剔除13名退出的参与者后,在122/669(18%)的表型匹配病例中,我们在排名靠前的基因中识别出了一个得到相位分析、ClinVar和ACMG分类支持的假定遗漏诊断。另外334/669(50%)的病例可能存在遗漏诊断,但需要功能验证。大规模应用GenePy为456/3183(14%)在隐性疾病基因中具有排名前5的GenePy分数的未诊断参与者识别出了潜在诊断,同时每个个体仅增加1.2个额外变异用于评估。