Ning Wenjuan, Marti Thomas M, Dorn Patrick, Peng Ren-Wang
Division of General Thoracic Surgery, Inselspital, Bern University Hospital, University of Bern, Bern, Switzerland.
Department for BioMedical Research (DBMR), University of Bern, Bern, Switzerland.
Front Oncol. 2022 Nov 22;12:1004669. doi: 10.3389/fonc.2022.1004669. eCollection 2022.
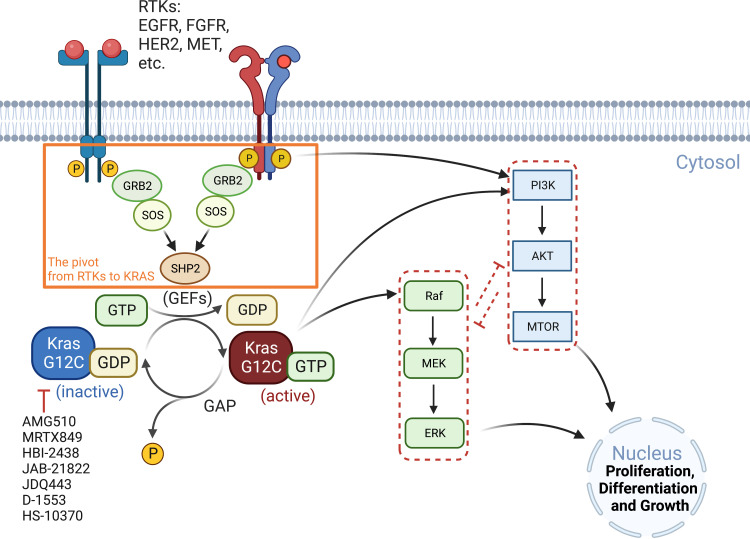
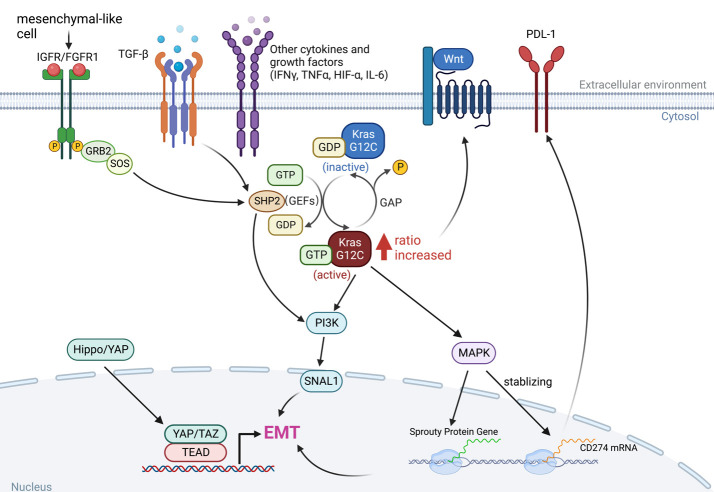
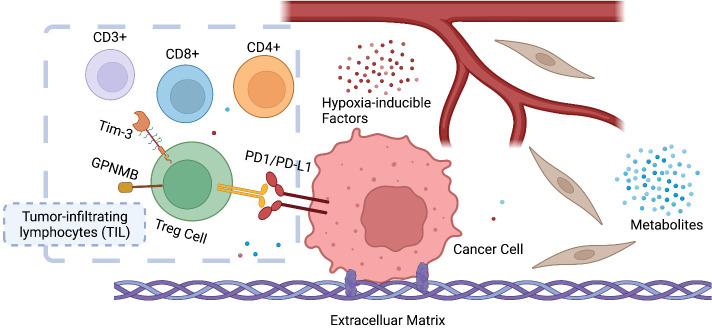
Adaptions to therapeutic pressures exerted on cancer cells enable malignant progression of the tumor, culminating in escape from programmed cell death and development of resistant diseases. A common form of cancer adaptation is non-genetic alterations that exploit mechanisms already present in cancer cells and do not require genetic modifications that can also lead to resistance mechanisms. Epithelial-to-mesenchymal transition (EMT) is one of the most prevalent mechanisms of adaptive drug resistance and resulting cancer treatment failure, driven by epigenetic reprogramming and EMT-specific transcription factors. A recent breakthrough in cancer treatment is the development of KRAS inhibitors, which herald a new era of therapy by knocking out a unique substitution of an oncogenic driver. However, these highly selective agents targeting KRAS, such as FDA-approved sotorasib (AMG510) and adagrasib (MRTX849), inevitably encounter multiple mechanisms of drug resistance. In addition to EMT, cancer cells can hijack or rewire the sophisticated signaling networks that physiologically control cell proliferation, growth, and differentiation to promote malignant cancer cell phenotypes, suggesting that inhibition of multiple interconnected signaling pathways may be required to block tumor progression on KRAS inhibitor therapy. Furthermore, the tumor microenvironment (TME) of cancer cells, such as tumor-infiltrating lymphocytes (TILs), contribute significantly to immune escape and tumor progression, suggesting a therapeutic approach that targets not only cancer cells but also the TME. Deciphering and targeting cancer adaptions promises mechanistic insights into tumor pathobiology and improved clinical management of KRAS-mutant cancer. This review presents recent advances in non-genetic adaptations leading to resistance to KRAS inhibitors, with a focus on oncogenic pathway rewiring, TME, and EMT.
对施加于癌细胞的治疗压力的适应会促使肿瘤恶性进展,最终导致癌细胞逃避程序性细胞死亡并发展出耐药性疾病。癌症适应的一种常见形式是非基因改变,这种改变利用癌细胞中已有的机制,不需要可能导致耐药机制的基因修饰。上皮-间质转化(EMT)是适应性耐药及癌症治疗失败最普遍的机制之一,由表观遗传重编程和EMT特异性转录因子驱动。癌症治疗的一项最新突破是KRAS抑制剂的研发,它通过消除致癌驱动因子的独特替代开启了一个新的治疗时代。然而,这些高度选择性的靶向KRAS的药物,如美国食品药品监督管理局(FDA)批准的索托拉西布(AMG510)和阿达格拉西布(MRTX849),不可避免地会遇到多种耐药机制。除了EMT,癌细胞还可以劫持或重新连接生理上控制细胞增殖、生长和分化的复杂信号网络,以促进恶性癌细胞表型,这表明在KRAS抑制剂治疗中可能需要抑制多个相互关联的信号通路来阻断肿瘤进展。此外,癌细胞的肿瘤微环境(TME),如肿瘤浸润淋巴细胞(TILs),对免疫逃逸和肿瘤进展有显著贡献,这表明一种不仅靶向癌细胞而且靶向TME的治疗方法。解读和靶向癌症适应有望为肿瘤病理生物学提供机制性见解,并改善KRAS突变型癌症的临床管理。本综述介绍了导致对KRAS抑制剂耐药的非基因适应的最新进展,重点关注致癌通路重编程、TME和EMT。