Genetics Section, Molecular and Clinical Sciences Research Institute, St. George's, University of London, London, United Kingdom.
Aging & Metabolism Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK.
Genet Med. 2023 Feb;25(2):100332. doi: 10.1016/j.gim.2022.11.001. Epub 2022 Dec 15.
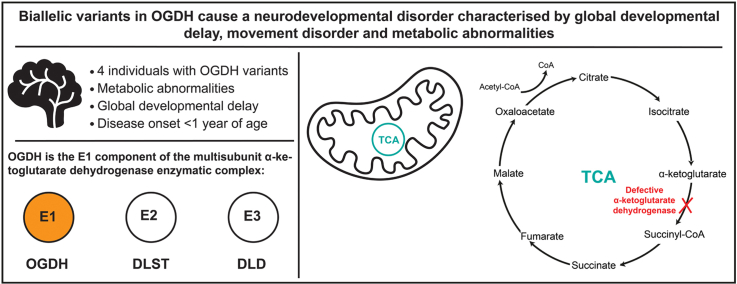
This study aimed to establish the genetic cause of a novel autosomal recessive neurodevelopmental disorder characterized by global developmental delay, movement disorder, and metabolic abnormalities.
We performed a detailed clinical characterization of 4 unrelated individuals from consanguineous families with a neurodevelopmental disorder. We used exome sequencing or targeted-exome sequencing, cosegregation, in silico protein modeling, and functional analyses of variants in HEK293 cells and Drosophila melanogaster, as well as in proband-derived fibroblast cells.
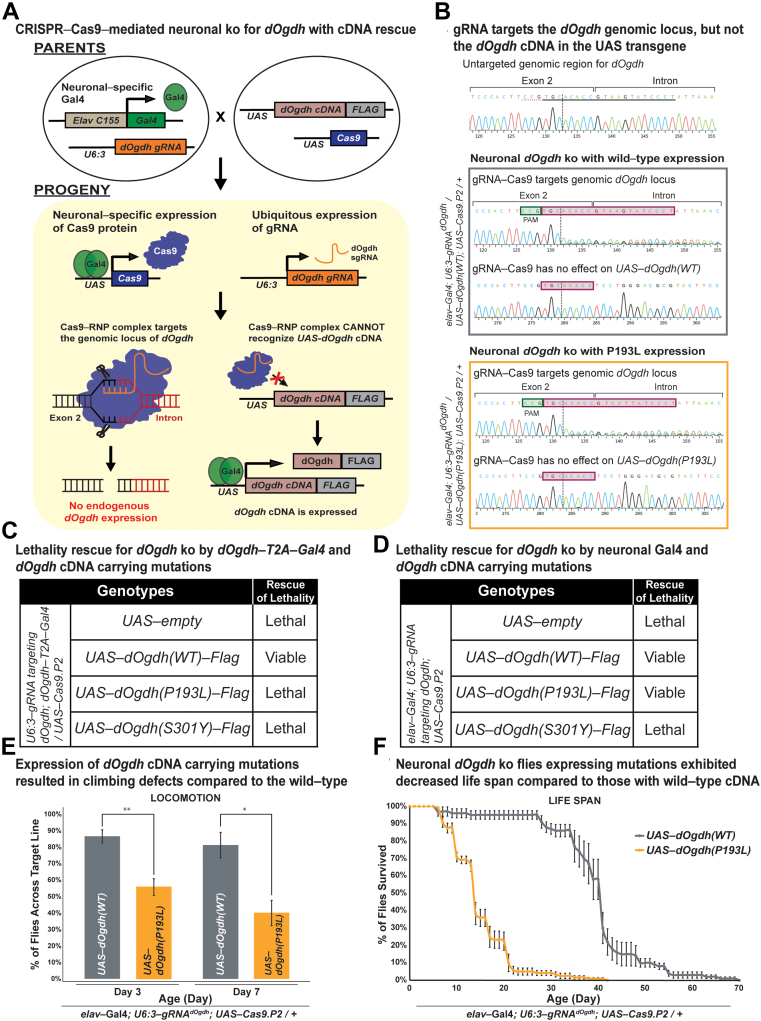
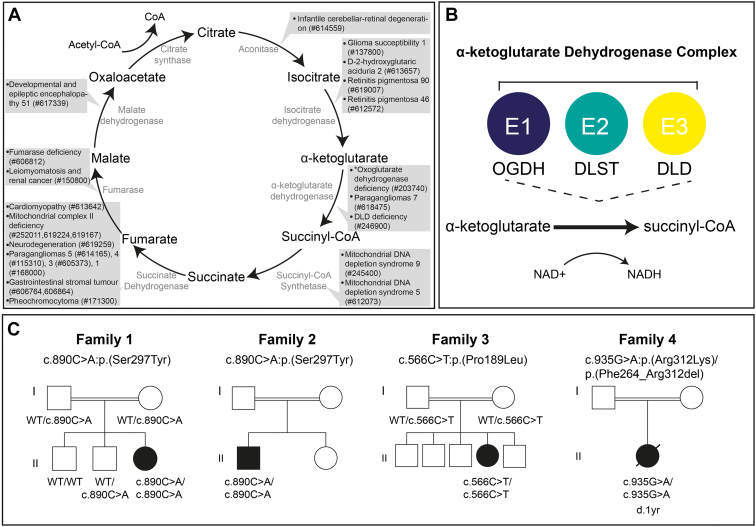
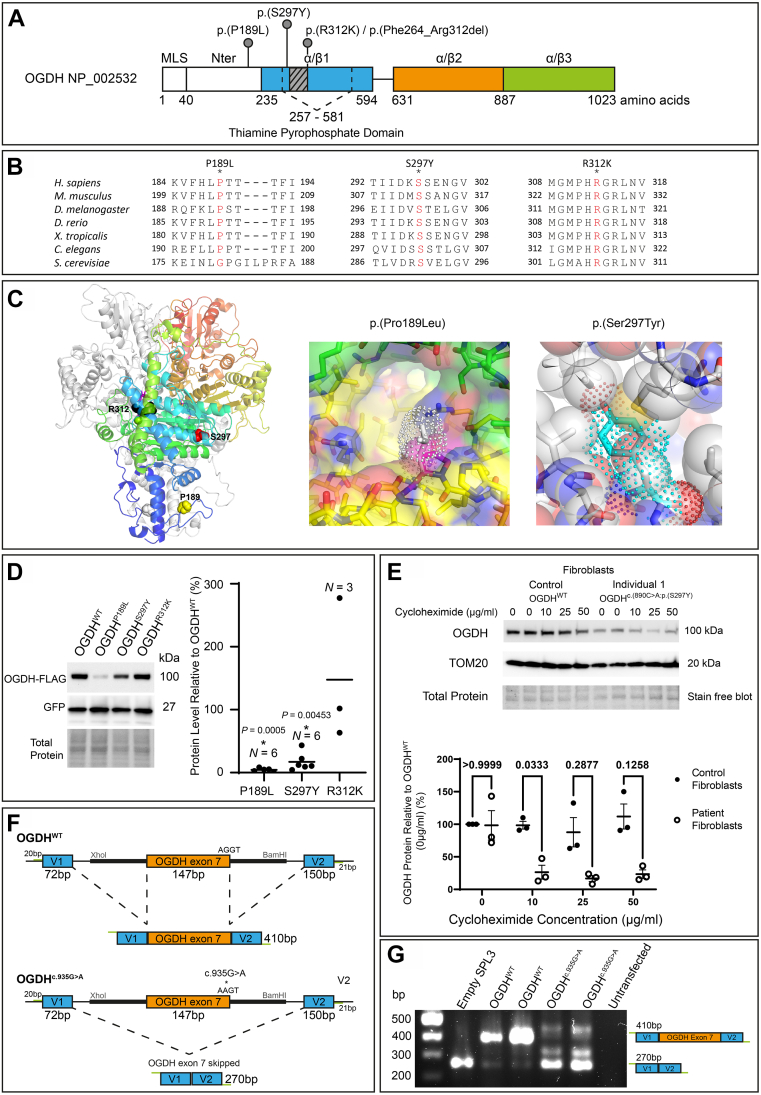
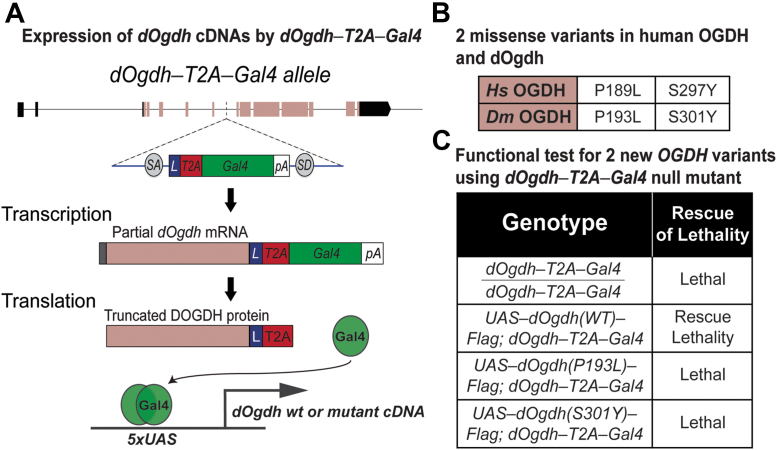
In the 4 individuals, we identified 3 novel homozygous variants in oxoglutarate dehydrogenase (OGDH) (NM_002541.3), which encodes a subunit of the tricarboxylic acid cycle enzyme α-ketoglutarate dehydrogenase. In silico homology modeling predicts that c.566C>T:p.(Pro189Leu) and c.890C>A:p.(Ser297Tyr) variants interfere with the structure and function of OGDH. Fibroblasts from individual 1 showed that the p.(Ser297Tyr) variant led to a higher degradation rate of the OGDH protein. OGDH protein with p.(Pro189Leu) or p.(Ser297Tyr) variants in HEK293 cells showed significantly lower levels than the wild-type protein. Furthermore, we showed that expression of Drosophila Ogdh (dOgdh) carrying variants homologous to p.(Pro189Leu) or p.(Ser297Tyr), failed to rescue developmental lethality caused by loss of dOgdh. SpliceAI, a variant splice predictor, predicted that the c.935G>A:p.(Arg312Lys)/p.(Phe264_Arg312del) variant impacts splicing, which was confirmed through a mini-gene assay in HEK293 cells.
We established that biallelic variants in OGDH cause a neurodevelopmental disorder with metabolic and movement abnormalities.
本研究旨在确定一种新的常染色体隐性神经发育障碍的遗传病因,该障碍的特征为全面发育迟缓、运动障碍和代谢异常。
我们对来自 4 个有神经发育障碍的近亲家庭的 4 个无关个体进行了详细的临床特征描述。我们使用外显子组测序或靶向外显子组测序、连锁分析、计算机模拟蛋白模型以及在 HEK293 细胞和黑腹果蝇以及来自先证者衍生的成纤维细胞中的变体的功能分析。
在这 4 个人中,我们发现了 3 种新的 OGDH(NM_002541.3)纯合变异,该基因编码三羧酸循环酶 α-酮戊二酸脱氢酶的一个亚基。计算机同源建模预测,c.566C>T:p.(Pro189Leu)和 c.890C>A:p.(Ser297Tyr)变异会干扰 OGDH 的结构和功能。个体 1 的成纤维细胞显示,p.(Ser297Tyr)变异导致 OGDH 蛋白的降解率更高。HEK293 细胞中的 OGDH 蛋白带有 p.(Pro189Leu)或 p.(Ser297Tyr)变异,其水平明显低于野生型蛋白。此外,我们表明,携带与 p.(Pro189Leu)或 p.(Ser297Tyr)同源的变体的 dOgdh 表达不能挽救 dOgdh 缺失引起的发育致死。变体剪接预测器 SpliceAI 预测 c.935G>A:p.(Arg312Lys)/p.(Phe264_Arg312del)变异会影响剪接,这在 HEK293 细胞中的微基因试验中得到了证实。
我们确定了 OGDH 的双等位基因变异导致具有代谢和运动异常的神经发育障碍。