Béquignon O J M, Bongers B J, Jespers W, IJzerman A P, van der Water B, van Westen G J P
Division of Drug Discovery and Safety, Leiden Academic Centre for Drug Research, Leiden University, Leiden, The Netherlands.
J Cheminform. 2023 Jan 6;15(1):3. doi: 10.1186/s13321-022-00672-x.
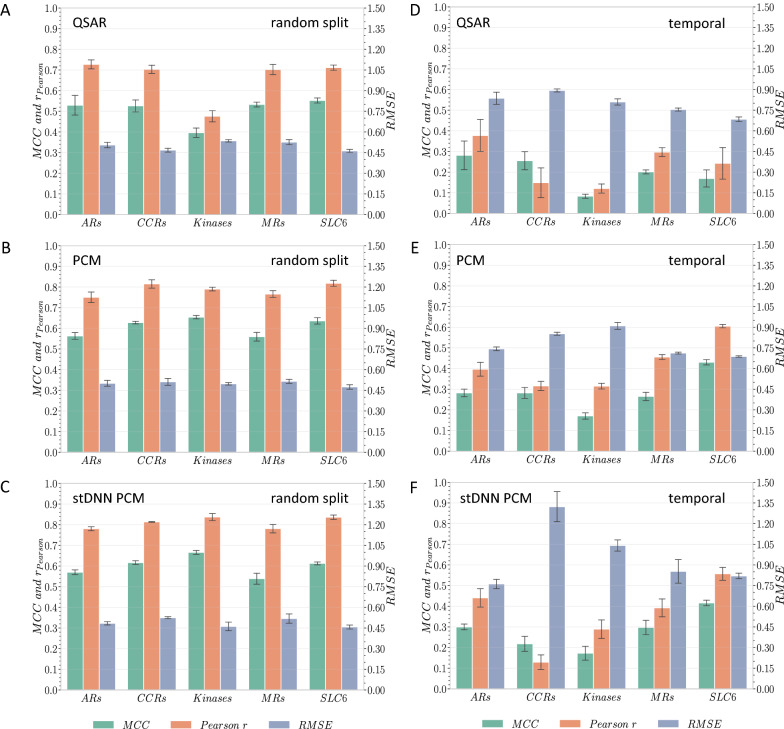
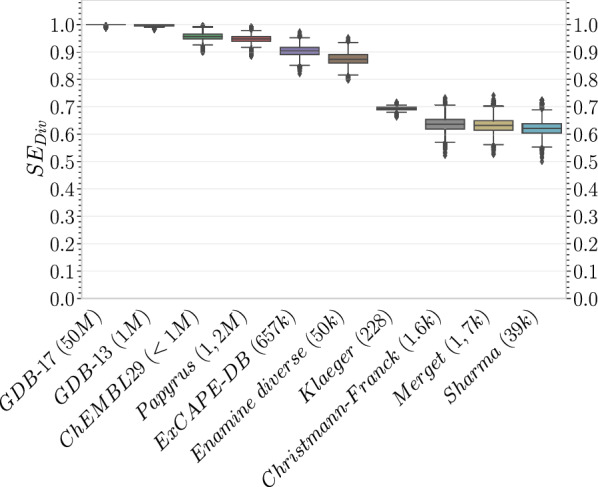
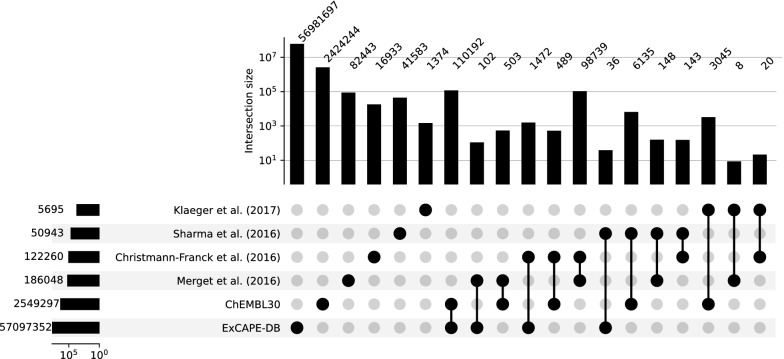
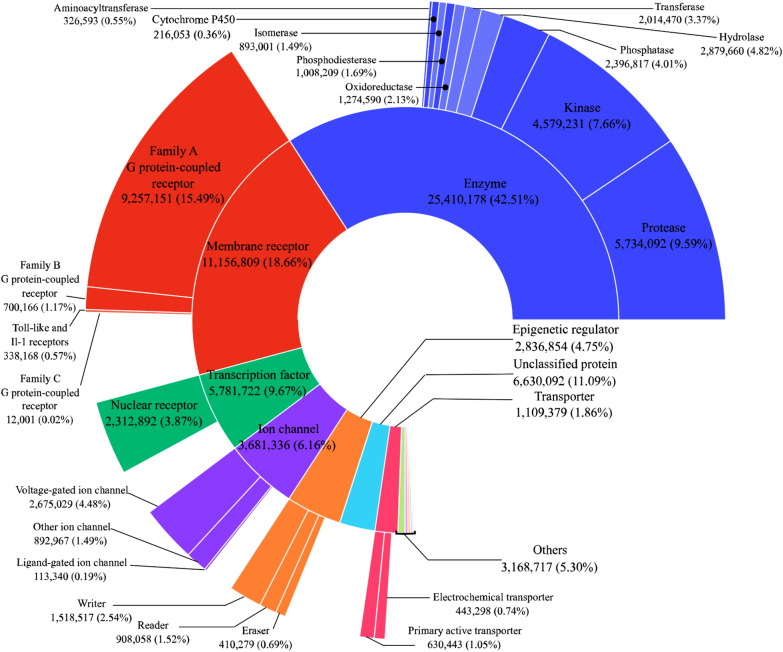
With the ongoing rapid growth of publicly available ligand-protein bioactivity data, there is a trove of valuable data that can be used to train a plethora of machine-learning algorithms. However, not all data is equal in terms of size and quality and a significant portion of researchers' time is needed to adapt the data to their needs. On top of that, finding the right data for a research question can often be a challenge on its own. To meet these challenges, we have constructed the Papyrus dataset. Papyrus is comprised of around 60 million data points. This dataset contains multiple large publicly available datasets such as ChEMBL and ExCAPE-DB combined with several smaller datasets containing high-quality data. The aggregated data has been standardised and normalised in a manner that is suitable for machine learning. We show how data can be filtered in a variety of ways and also perform some examples of quantitative structure-activity relationship analyses and proteochemometric modelling. Our ambition is that this pruned data collection constitutes a benchmark set that can be used for constructing predictive models, while also providing an accessible data source for research.
随着公开可用的配体-蛋白质生物活性数据持续快速增长,有大量宝贵数据可用于训练众多机器学习算法。然而,并非所有数据在规模和质量上都是等同的,研究人员需要花费大量时间来使数据符合他们的需求。除此之外,为一个研究问题找到合适的数据本身往往就是一项挑战。为应对这些挑战,我们构建了纸莎草纸数据集。纸莎草纸数据集由大约6000万个数据点组成。该数据集包含多个大型公开可用数据集,如ChEMBL和ExCAPE-DB,还结合了几个包含高质量数据的较小数据集。汇总后的数据已以适合机器学习的方式进行了标准化和归一化处理。我们展示了如何以多种方式对数据进行筛选,还进行了一些定量构效关系分析和蛋白质化学计量学建模的示例。我们的目标是,这个经过精简的数据集合构成一个可用于构建预测模型的基准集,同时也为研究提供一个易于获取的数据源。