Thomas Morgan, Smith Robert T, O'Boyle Noel M, de Graaf Chris, Bender Andreas
Centre for Molecular Informatics, Department of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK.
Computational Chemistry, Sosei Heptares, Steinmetz Building, Granta Park, Great Abington, Cambridge, CB21 6DG, UK.
J Cheminform. 2021 May 13;13(1):39. doi: 10.1186/s13321-021-00516-0.
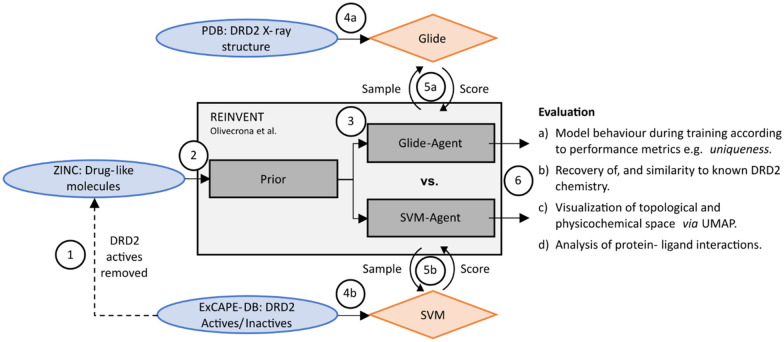
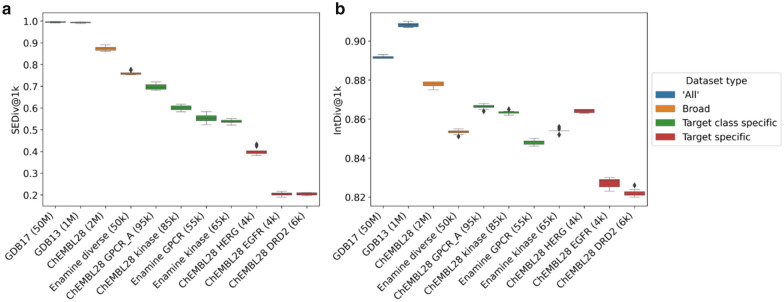
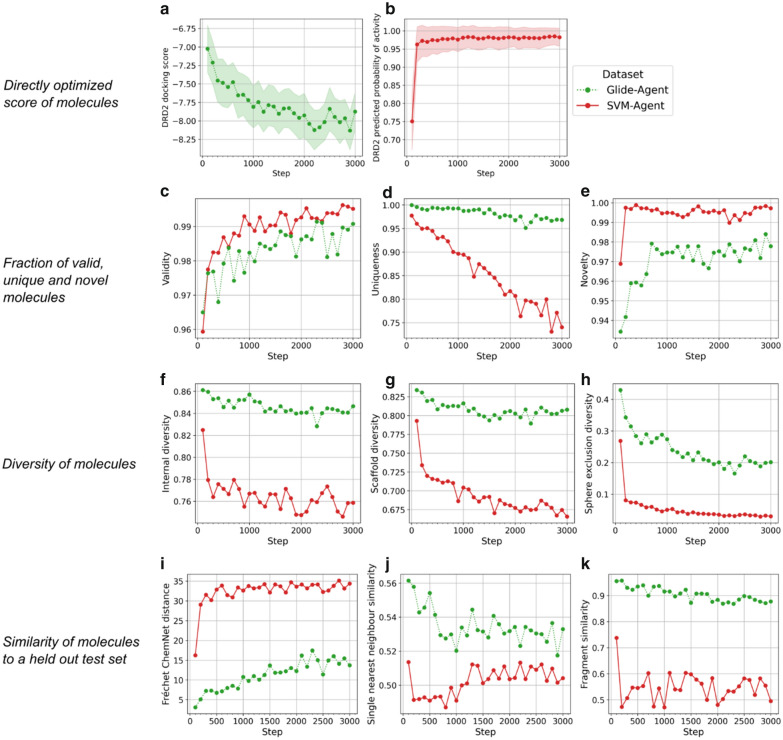
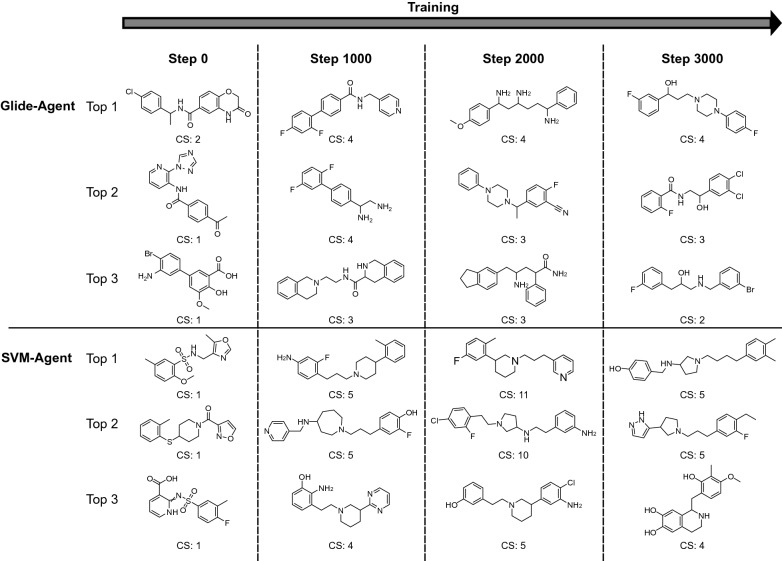
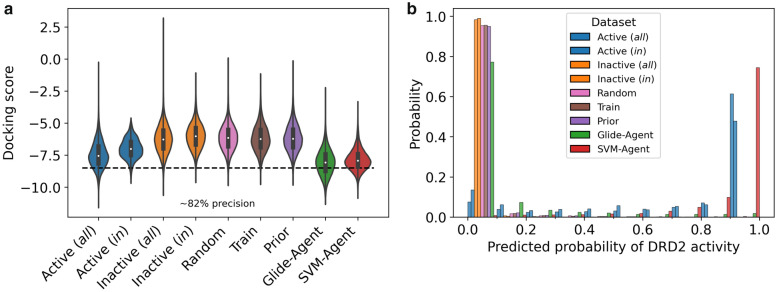
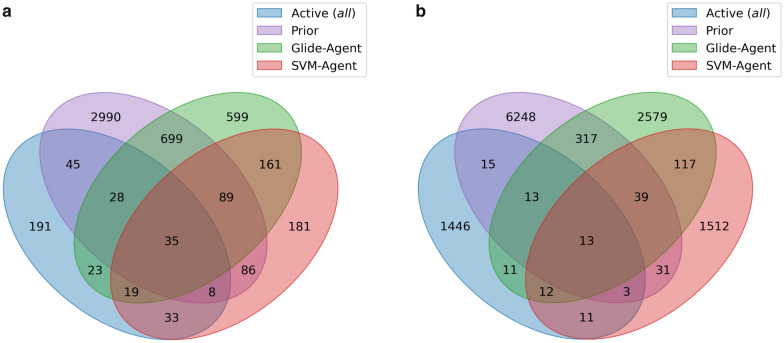
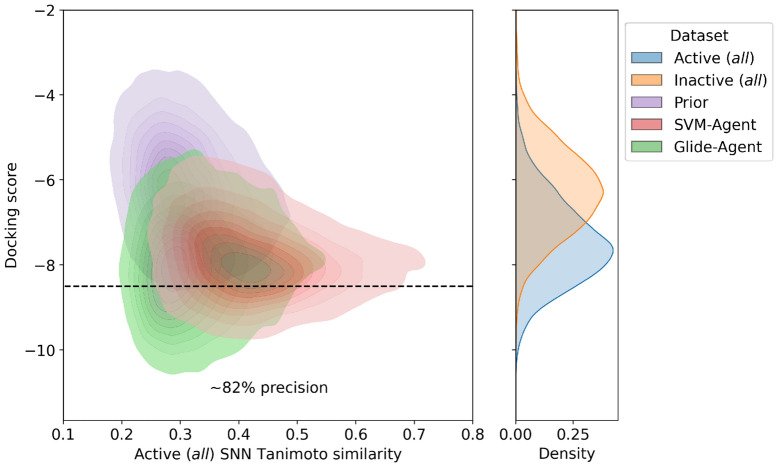
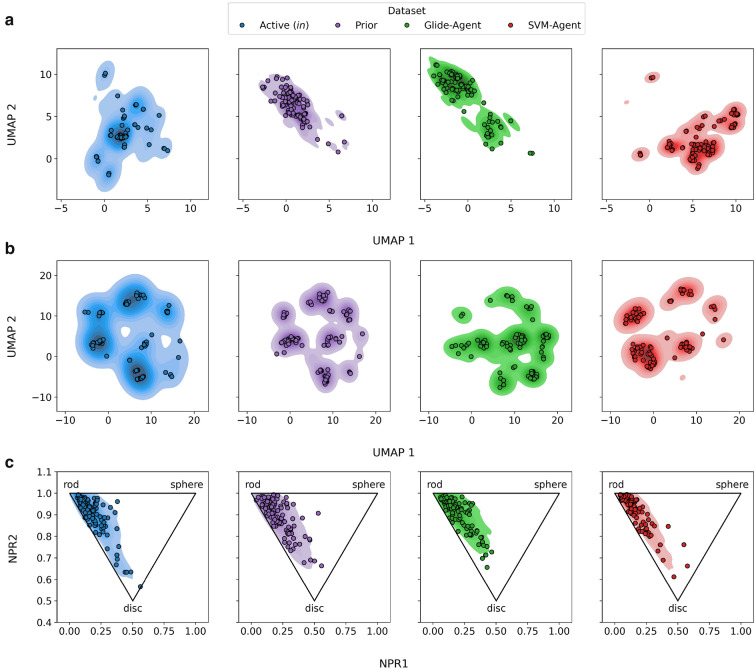
Deep generative models have shown the ability to devise both valid and novel chemistry, which could significantly accelerate the identification of bioactive compounds. Many current models, however, use molecular descriptors or ligand-based predictive methods to guide molecule generation towards a desirable property space. This restricts their application to relatively data-rich targets, neglecting those where little data is available to sufficiently train a predictor. Moreover, ligand-based approaches often bias molecule generation towards previously established chemical space, thereby limiting their ability to identify truly novel chemotypes. In this work, we assess the ability of using molecular docking via Glide-a structure-based approach-as a scoring function to guide the deep generative model REINVENT and compare model performance and behaviour to a ligand-based scoring function. Additionally, we modify the previously published MOSES benchmarking dataset to remove any induced bias towards non-protonatable groups. We also propose a new metric to measure dataset diversity, which is less confounded by the distribution of heavy atom count than the commonly used internal diversity metric. With respect to the main findings, we found that when optimizing the docking score against DRD2, the model improves predicted ligand affinity beyond that of known DRD2 active molecules. In addition, generated molecules occupy complementary chemical and physicochemical space compared to the ligand-based approach, and novel physicochemical space compared to known DRD2 active molecules. Furthermore, the structure-based approach learns to generate molecules that satisfy crucial residue interactions, which is information only available when taking protein structure into account. Overall, this work demonstrates the advantage of using molecular docking to guide de novo molecule generation over ligand-based predictors with respect to predicted affinity, novelty, and the ability to identify key interactions between ligand and protein target. Practically, this approach has applications in early hit generation campaigns to enrich a virtual library towards a particular target, and also in novelty-focused projects, where de novo molecule generation either has no prior ligand knowledge available or should not be biased by it.
深度生成模型已展现出设计有效且新颖化学结构的能力,这能够显著加速生物活性化合物的识别。然而,当前许多模型使用分子描述符或基于配体的预测方法,将分子生成引导至理想的性质空间。这限制了它们在相对数据丰富的靶点上的应用,而忽略了那些几乎没有足够数据来充分训练预测器的靶点。此外,基于配体的方法通常会使分子生成偏向于先前已确立的化学空间,从而限制了它们识别真正新颖化学型的能力。在这项工作中,我们评估了通过Glide(一种基于结构的方法)进行分子对接作为评分函数来引导深度生成模型REINVENT的能力,并将模型性能和行为与基于配体的评分函数进行比较。此外,我们修改了先前发布的MOSES基准数据集,以消除对不可质子化基团的任何诱导偏差。我们还提出了一种新的指标来衡量数据集的多样性,与常用的内部多样性指标相比,该指标受重原子计数分布的干扰较小。关于主要发现,我们发现当针对DRD2优化对接分数时,该模型提高了预测的配体亲和力,超过了已知的DRD2活性分子。此外,与基于配体的方法相比,生成的分子占据互补的化学和物理化学空间,与已知的DRD2活性分子相比,占据新颖的物理化学空间。此外,基于结构的方法学会生成满足关键残基相互作用的分子,而这种信息只有在考虑蛋白质结构时才会出现。总体而言,这项工作证明了在预测亲和力、新颖性以及识别配体与蛋白质靶点之间关键相互作用的能力方面,使用分子对接来引导从头分子生成优于基于配体的预测器。实际上,这种方法可应用于早期命中化合物生成活动,以针对特定靶点丰富虚拟库,也可应用于注重新颖性的项目,在这些项目中,从头分子生成要么没有可用的先前配体知识,要么不应受其影响。