Faculty of Infectious and Tropical Diseases, London School of Hygiene & Tropical Medicine, London, UK.
Research Institute for Medicines (iMed.ULisboa), Faculdade de Farmácia, Universidade de Lisboa, Lisboa, Portugal.
Genome Med. 2023 Jan 19;15(1):3. doi: 10.1186/s13073-023-01153-y.
Klebsiella pneumoniae (Kp) Gram-negative bacteria cause nosocomial infections and rapidly acquire antimicrobial resistance (AMR), which makes it a global threat to human health. It also has a comparatively rare hypervirulent phenotype that can lead to severe disease in otherwise healthy individuals. Unlike classic Kp, canonical hypervirulent strains usually have limited AMR. However, after initial case reports in 2015, carbapenem-resistant hypervirulent Kp has increased in prevalence, including in China, but there is limited understanding of its burden in other geographical regions.
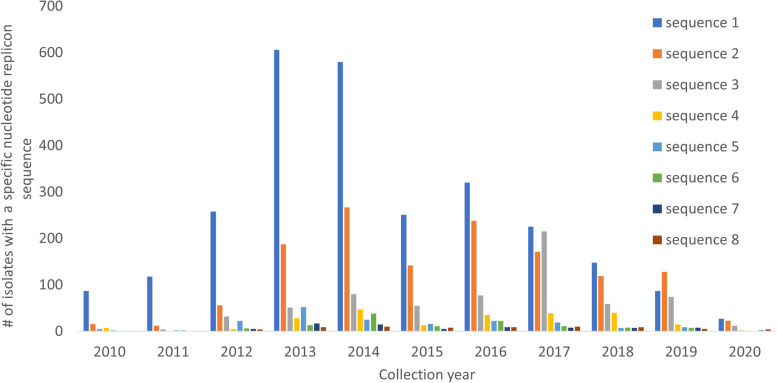
Here, we examined the largest collection of publicly available sequenced Kp isolates (n=13,178), containing 1603 different sequence types (e.g. ST11 15.0%, ST258 9.5%), and 2174 (16.5%) hypervirulent strains. We analysed the plasmid replicons and carbapenemase and siderophore encoding genes to understand the movement of hypervirulence and AMR genes located on plasmids, and their convergence in carbapenem-resistant hypervirulent Kp.
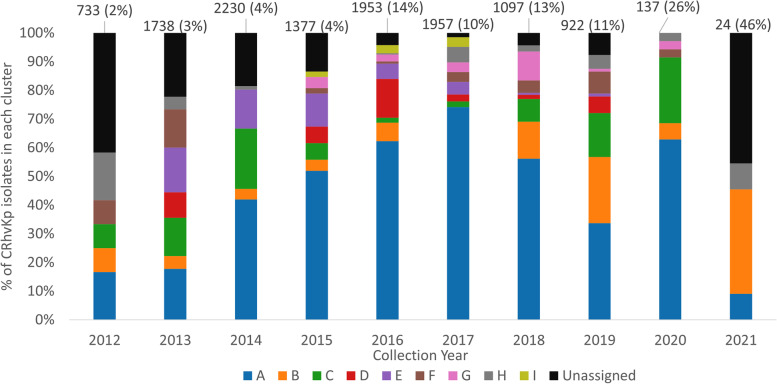
We identified and analysed 3034 unique plasmid replicons to inform the epidemiology and transmission dynamics of carbapenem-resistant hypervirulent Kp (n=1028, 7.8%). We found several outbreaks globally, including one involving ST11 strains in China and another of ST231 in Asia centred on India, Thailand, and Pakistan. There was evidence of global flow of Kp, including across multiple continents. In most cases, clusters of Kp isolates are the result of hypervirulence genes entering classic strains, instead of carbapenem resistance genes entering canonical hypervirulent ones.
Our analysis demonstrates the importance of plasmid analysis in the monitoring of carbapenem-resistant and hypervirulent strains of Kp. With the growing adoption of omics-based technologies for clinical and surveillance applications, including in geographical regions with gaps in data and knowledge (e.g. sub-Saharan Africa), the identification of the spread of AMR will inform infection control globally.
肺炎克雷伯菌(Kp)革兰氏阴性细菌引起医院感染,并迅速获得抗生素耐药性(AMR),这对全球人类健康构成威胁。它还具有一种相对罕见的高毒力表型,可导致原本健康的个体患上严重疾病。与经典 Kp 不同,典型的高毒力菌株通常具有有限的 AMR。然而,自 2015 年首次病例报告以来,耐碳青霉烯类高毒力 Kp 的流行率有所增加,包括在中国,但对其他地理区域的负担了解有限。
在这里,我们检查了最大的公开可用测序 Kp 分离株集合(n=13,178),其中包含 1603 种不同的序列类型(例如 ST11,15.0%;ST258,9.5%)和 2174 种(16.5%)高毒力菌株。我们分析了质粒复制子和碳青霉烯酶和铁载体编码基因,以了解位于质粒上的高毒力和 AMR 基因的运动及其在耐碳青霉烯类高毒力 Kp 中的趋同。
我们鉴定和分析了 3034 个独特的质粒复制子,以了解耐碳青霉烯类高毒力 Kp 的流行病学和传播动态(n=1028,7.8%)。我们发现了一些全球范围内的暴发,包括中国的 ST11 株和以印度、泰国和巴基斯坦为中心的亚洲的 ST231 株。有证据表明 Kp 在全球范围内流动,包括跨越多个大陆。在大多数情况下,Kp 分离株的聚类是高毒力基因进入经典菌株的结果,而不是碳青霉烯类耐药基因进入典型高毒力菌株的结果。
我们的分析表明质粒分析在监测耐碳青霉烯类和高毒力 Kp 菌株中的重要性。随着基于组学的技术在临床和监测应用中的日益普及,包括在数据和知识存在差距的地理区域(例如撒哈拉以南非洲),对 AMR 传播的识别将在全球范围内为感染控制提供信息。