Department of Medical Oncology, Cochin Hospital, Paris Cancer Institute CARPEM, Université Paris Cité, APHP.Centre, 75014 Paris, France.
INSERM U1016-CNRS UMR8104, Cochin Institute, Paris Cancer Institute CARPEM, Université Paris Cité, APHP.Centre, 75014 Paris, France.
Int J Mol Sci. 2023 Jan 10;24(2):1361. doi: 10.3390/ijms24021361.
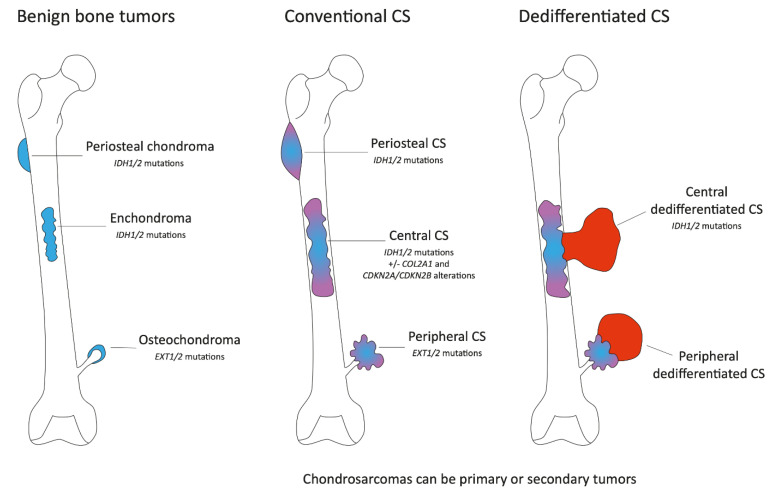
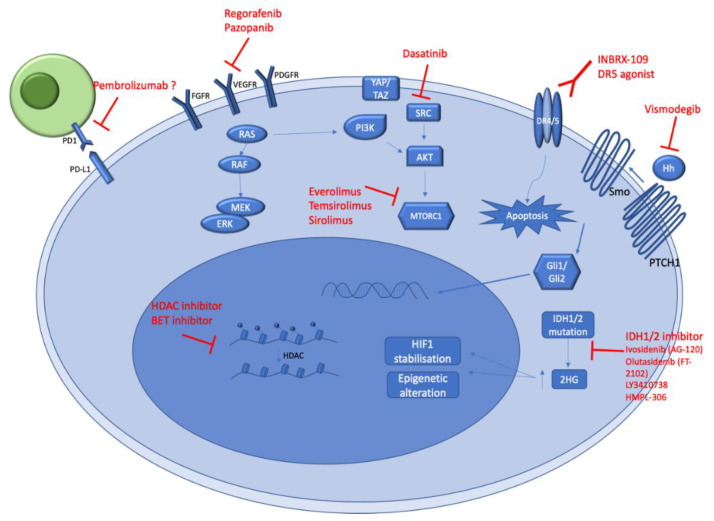
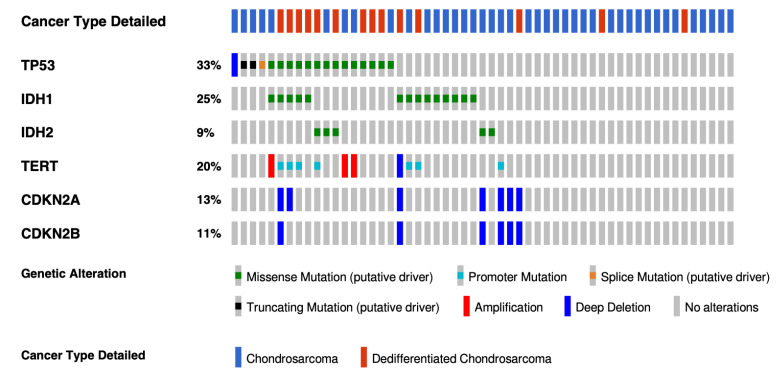
This review provides an overview of histopathology, clinical presentation, molecular pathways, and potential new systemic treatments of high-grade chondrosarcomas (CS), including grade 2−3 conventional, dedifferentiated, and mesenchymal CS. The diagnosis of CS combines radiological and histological data in conjunction with patient clinical presentations. Conventional CS is the most frequent subtype of CS (85%) and represents about 25% of primary bone tumors in adults; they can be categorized according to their bone location into central, peripheral, and periosteal chondrosarcomas. Central and peripheral CS differ at the molecular level with either IDH1/2 mutations or EXT1/2 mutations, respectively. CDKN2A/B deletions are also frequent in conventional CS, as well as COL2A1 mutations. Dedifferentiated CS develops when low-grade conventional CS transforms into a high-grade sarcoma and most frequently exhibits features of osteosarcoma, fibrosarcoma, or undifferentiated pleomorphic sarcoma. Their molecular characteristics are similar to conventional CS. Mesenchymal CS is a totally different pathological entity exhibiting recurrent translocations. Their clinical presentation and management are different too. The standard treatment of CSs is wide en-bloc resection. CS are relatively radiotherapy resistant; therefore, doses >60 Gy are needed in an attempt to achieve local control in unresectable tumors. Chemotherapy is possibly effective in mesenchymal chondrosarcoma and is of uncertain value in dedifferentiated chondrosarcoma. Due to resistance to standard anticancer agents, the prognosis is poor in patients with metastatic or unresectable chondrosarcomas. Recently, the refined characterization of the molecular profile, as well as the development of new treatments, allow new therapeutic options for these rare tumors. The efficiency of IDH1 inhibitors in other malignancies suggests that these inhibitors will be part of IDH1/2 mutated conventional CS management soon. Other treatment approaches, such as PIK3-AKT-mTOR inhibitors, cell cycle inhibitors, and epigenetic or immune modulators based on improving our understanding of CS molecular biology, are emerging.
这篇综述概述了高级别软骨肉瘤(CS)的组织病理学、临床表现、分子途径以及潜在的新系统治疗方法,包括 2-3 级常规、去分化和间充质 CS。CS 的诊断结合了影像学和组织学数据以及患者的临床表现。常规 CS 是 CS 中最常见的亚型(85%),占成人原发性骨肿瘤的 25%左右;它们可以根据骨的位置分为中央、周围和骨膜 CS。中央和周围 CS 在分子水平上有所不同,分别存在 IDH1/2 突变或 EXT1/2 突变。CDKN2A/B 缺失在常规 CS 中也很常见,以及 COL2A1 突变。低级别常规 CS 转化为高级别肉瘤时会发生去分化 CS,最常表现为骨肉瘤、纤维肉瘤或未分化多形性肉瘤的特征。它们的分子特征与常规 CS 相似。间充质 CS 是一种完全不同的病理实体,表现出复发性易位。它们的临床表现和治疗也不同。CS 的标准治疗方法是广泛整块切除。CS 对放疗相对耐药;因此,需要 >60Gy 的剂量试图在不可切除的肿瘤中实现局部控制。化疗可能对间充质软骨肉瘤有效,对去分化软骨肉瘤的价值不确定。由于对标准抗癌药物的耐药性,转移性或不可切除的软骨肉瘤患者预后较差。最近,对分子特征的精细描述以及新治疗方法的开发,为这些罕见肿瘤提供了新的治疗选择。IDH1 抑制剂在其他恶性肿瘤中的有效性表明,这些抑制剂很快将成为 IDH1/2 突变的常规 CS 治疗的一部分。其他治疗方法,如 PI3K-AKT-mTOR 抑制剂、细胞周期抑制剂以及基于改善我们对 CS 分子生物学理解的表观遗传或免疫调节剂,正在出现。