Shang Shuai, Zhang Zaiwang, Zhao Liping, Liu Longxiang, Shi Dongli, Xu Hui, Zhang Hanjie, Xie Wenjun, Zhao Fengjuan, Zhou Zhihao, Xu Jikun, Wang Jun
School of Biological and Environmental Engineering, Binzhou University, Binzhou 256600, China.
School of Environmental & Municipal Engineering, Qingdao University of Technology, Qingdao 266000, China.
Microorganisms. 2022 Dec 21;11(1):18. doi: 10.3390/microorganisms11010018.
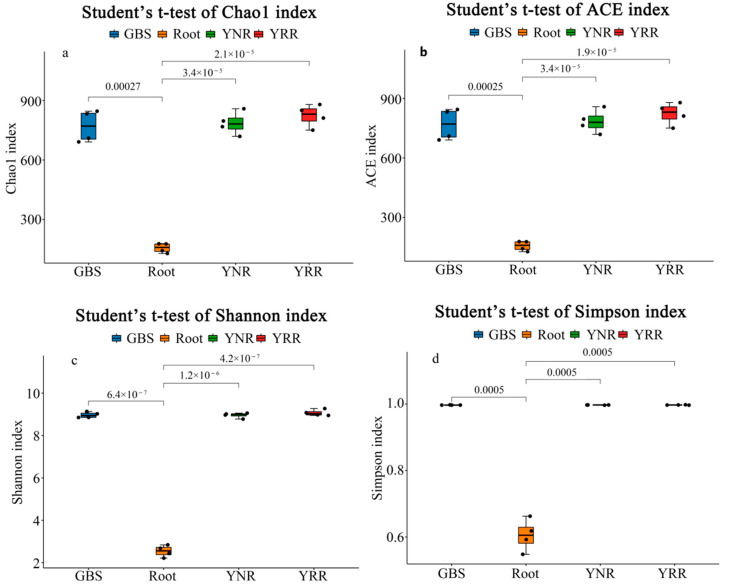
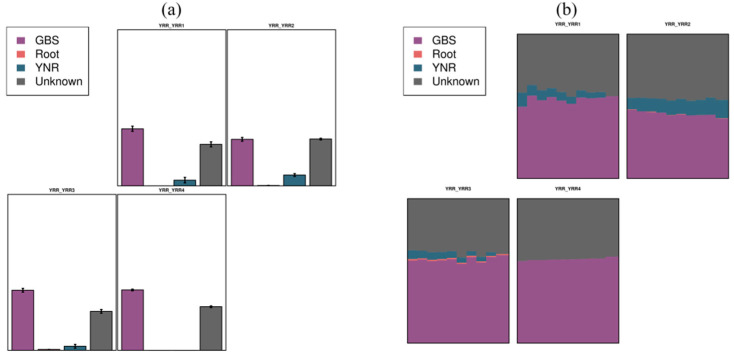
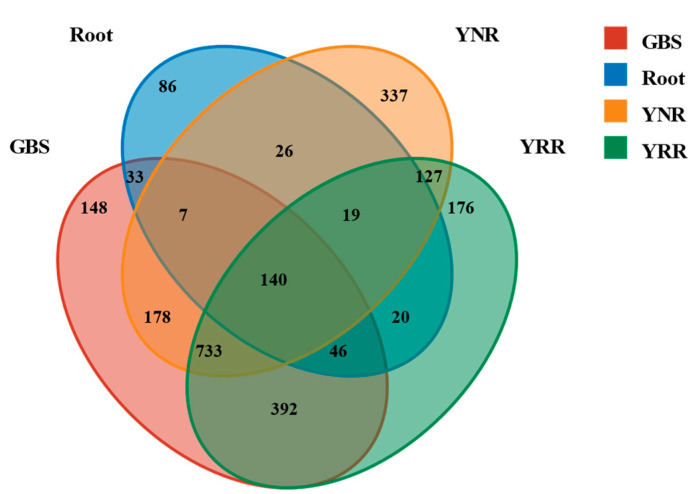
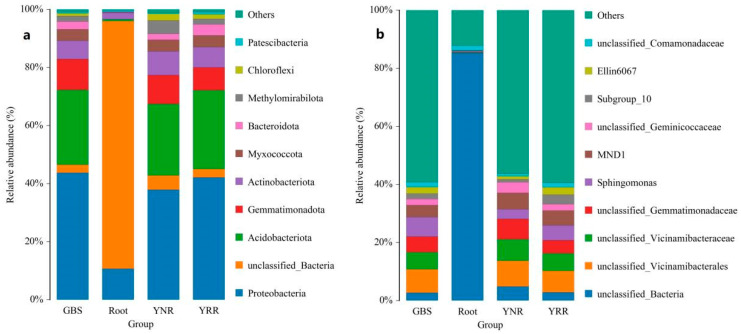
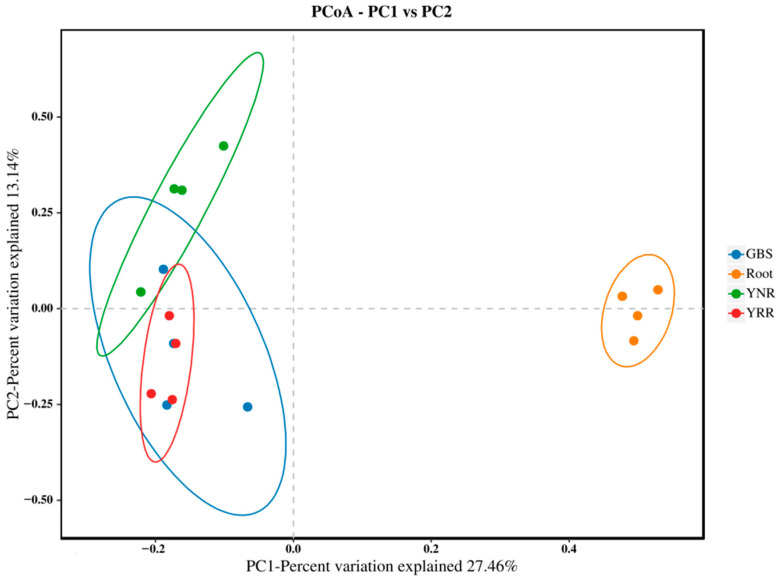
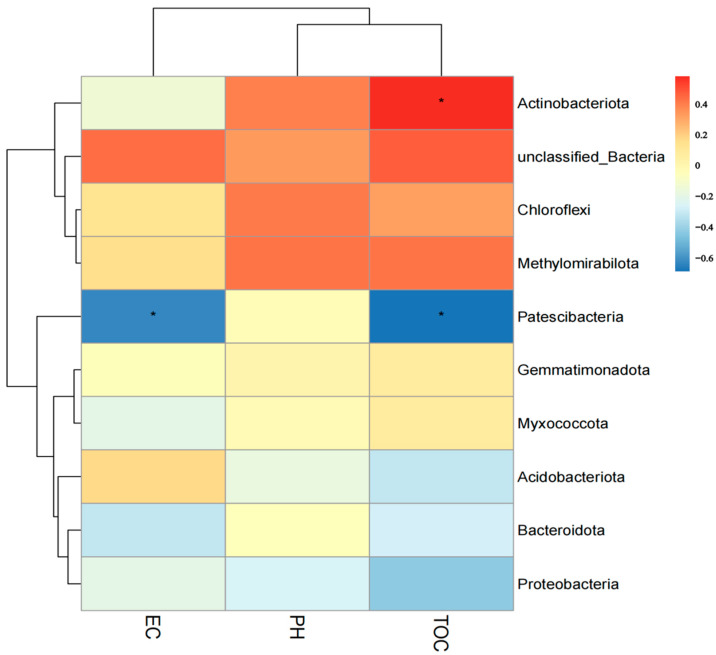
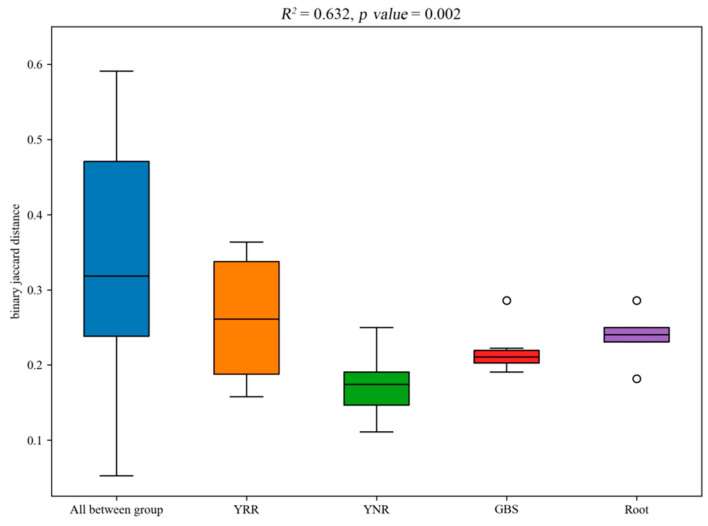
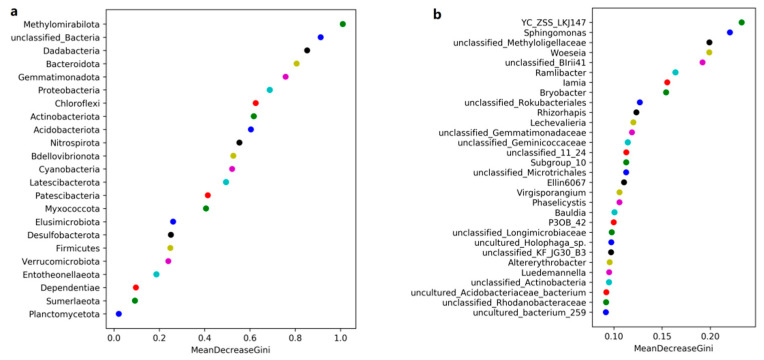
L., as an invasive plant, has negatively impacted the ecosystem functioning and stability of the terrestrial ecosystem in China. However, little information was available for its effects on microorganisms in the Yellow River Delta (YRD), the biggest newly-formed wetland in China. In the present study, high-throughput sequencing technology was used to obtain the bacterial community in soils and roots of different plant species, including and some native ones in the YRD. Our results showed that the Proteobacteria, Acidobacteriota, Gemmatimonadota, and Actinobacteriota were dominant in the rhizosphere soils of (84.2%) and (86.47%), and the bulk soils (80.7%). The Proteobacteria and Actinobacteriota were dominant within the root of . A total of 2468 bacterial OTUs were obtained from different groups among which 140 were observed in all the groups; 1019 OTUs were shared by non-rhizosphere soil bacteria (YNR) rhizosphere soil bacteria (YRR) groups. The indexes of the ACE (823.1), Chao1 (823.19), Simpson (0.9971), and Shannon (9.068) were the highest in the YRR groups, showing the greatest bacterial community diversity. Random forest analysis showed that the Methylomirabilota and Dadabacteria (at the phylum level) and the , and (at the genus level) were identified as the main predictors among different groups. The LEfSe results also showed the essential role of the Acidobacteriota in the YRR group. The SourceTracker analysis of the bacterial community of the YRR group was mainly from GBS groups (average 53.14%) and a small part was from YNR groups (average 6.56%), indicating that the invasion had a more significant effect on native plants' rhizosphere microorganisms than soil microorganisms. Our observations could provide valuable information for understanding the bacterial diversity and structure of the soil to the invasion of .
作为一种入侵植物,[植物名称未给出]对中国陆地生态系统的生态系统功能和稳定性产生了负面影响。然而,关于其对中国最大的新形成湿地——黄河三角洲(YRD)微生物的影响,目前几乎没有相关信息。在本研究中,利用高通量测序技术获取了黄河三角洲不同植物物种(包括[植物名称未给出]和一些本地植物)土壤和根系中的细菌群落。我们的结果表明,变形菌门、酸杆菌门、芽单胞菌门和放线菌门在[植物名称未给出](84.2%)和[植物名称未给出](86.47%)的根际土壤以及大块土壤(80.7%)中占主导地位。变形菌门和放线菌门在[植物名称未给出]的根系中占主导地位。从不同组中共获得2468个细菌OTU,其中140个在所有组中都有观察到;1019个OTU由非根际土壤细菌(YNR)和根际土壤细菌(YRR)组共享。ACE(823.1)、Chao1(823.19)、辛普森(0.9971)和香农(9.068)指数在YRR组中最高,表明细菌群落多样性最大。随机森林分析表明,甲基微菌门和达德菌门(在门水平)以及[属名称未给出]、[属名称未给出]和[属名称未给出](在属水平)被确定为不同组中的主要预测因子。LEfSe结果还表明酸杆菌门在YRR组中的重要作用。YRR组细菌群落的SourceTracker分析主要来自GBS组(平均53.14%),一小部分来自YNR组(平均6.56%),这表明[植物名称未给出]入侵对本地植物根际微生物的影响比对土壤微生物的影响更大。我们的观察结果可为了解[植物名称未给出]入侵对土壤细菌多样性和结构的影响提供有价值的信息。