Sugisaki Kenji, Toyota Kazuo, Sato Kazunobu, Shiomi Daisuke, Takui Takeji
Department of Chemistry and Molecular Materials Science, Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka, 558-8585, Japan.
JST PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama, 332-0012, Japan.
Commun Chem. 2022 Jul 25;5(1):84. doi: 10.1038/s42004-022-00701-8.
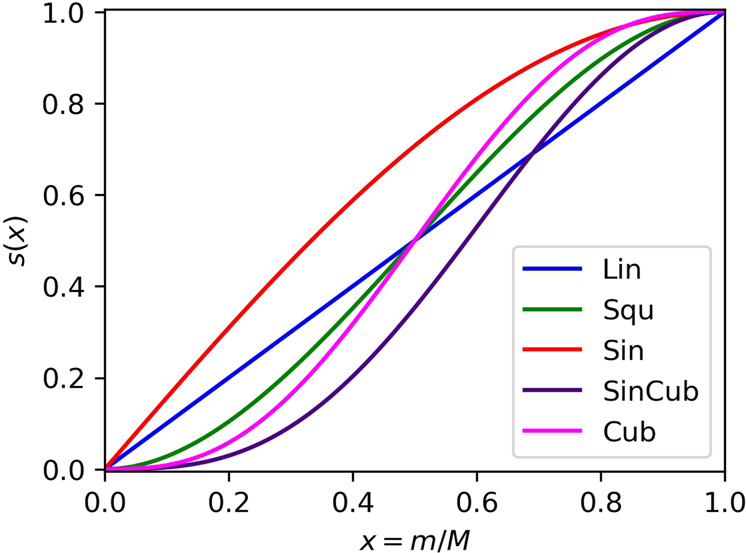
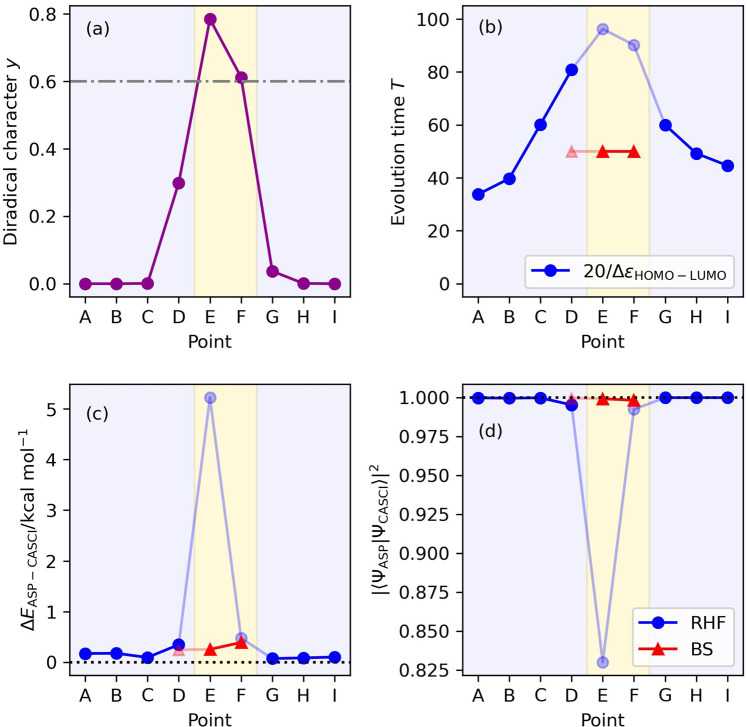
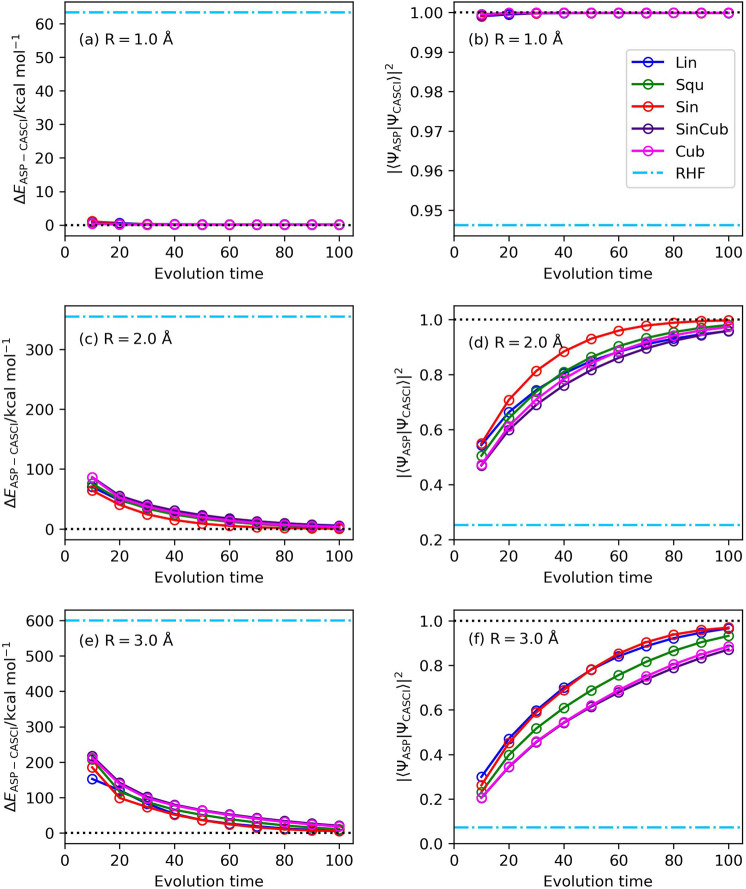
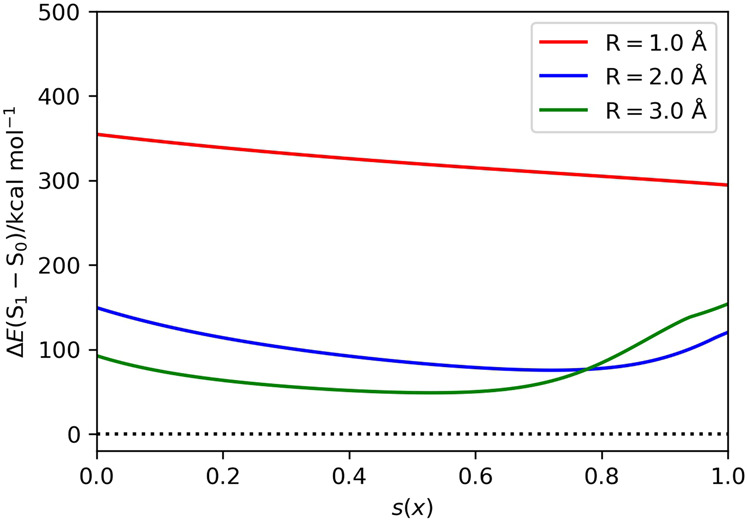
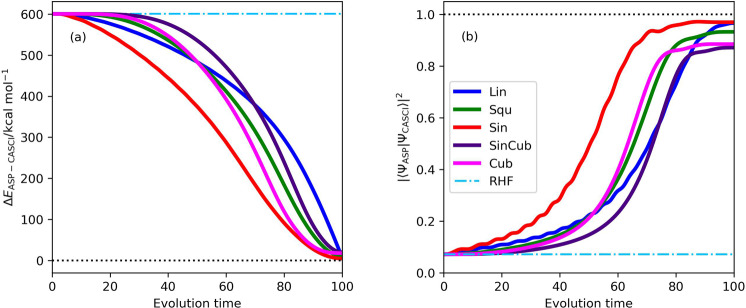
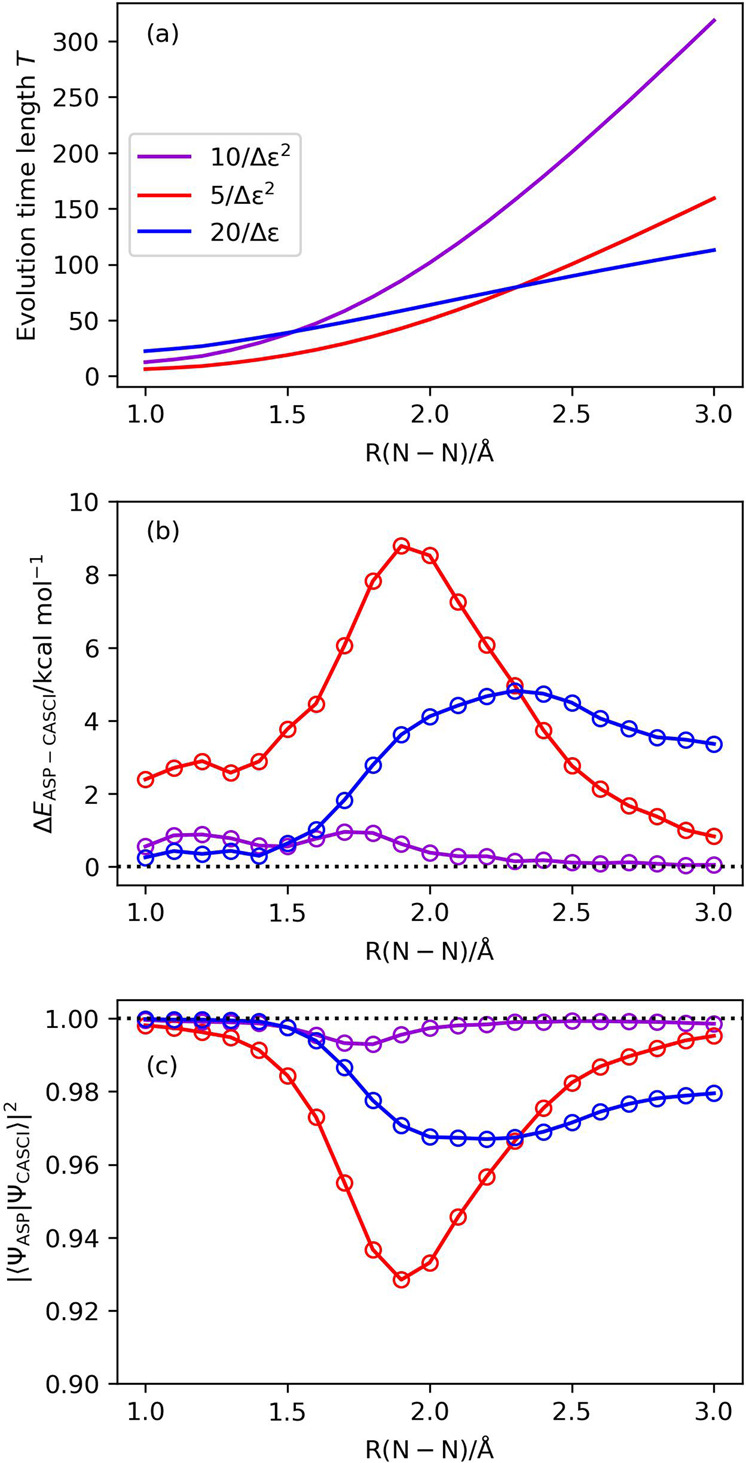
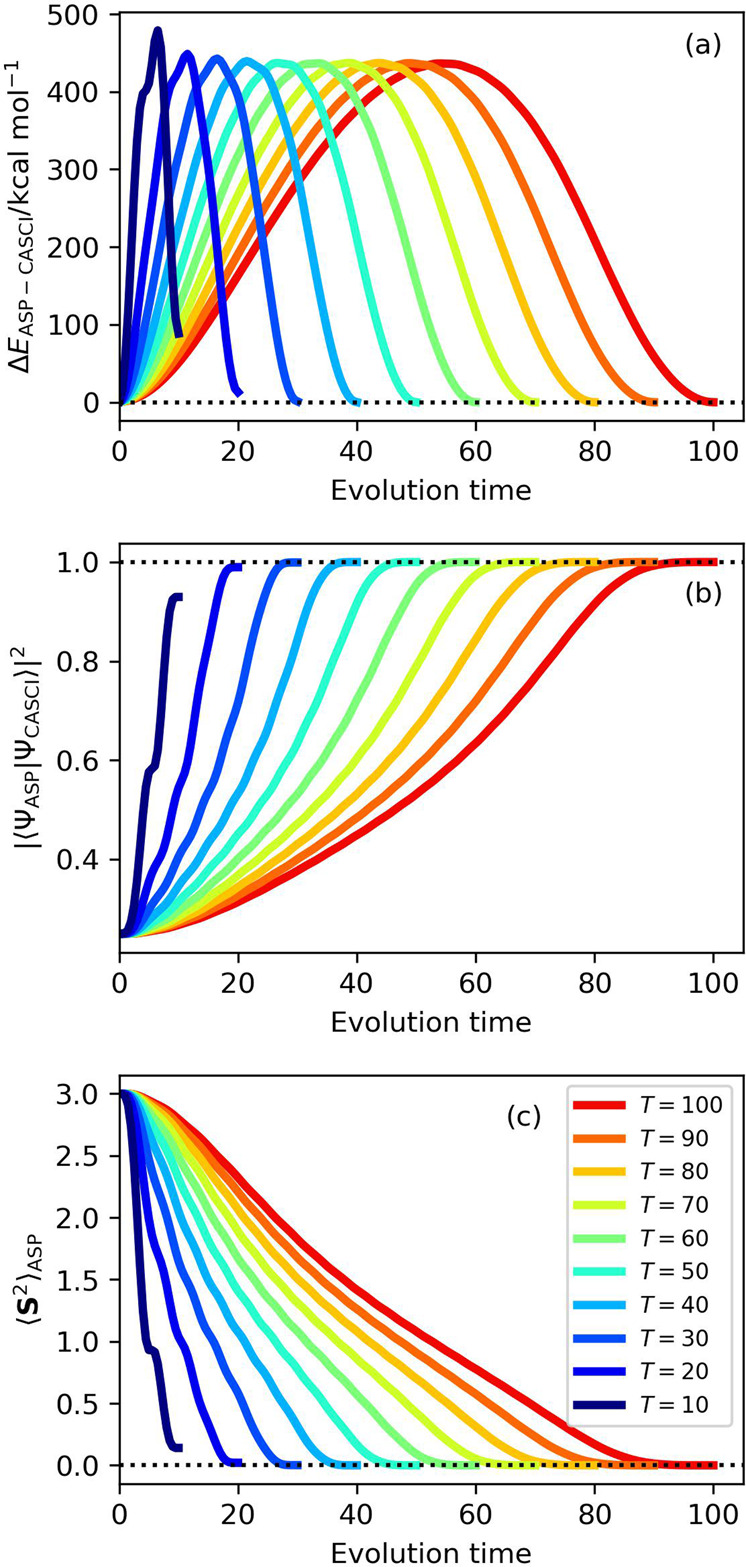
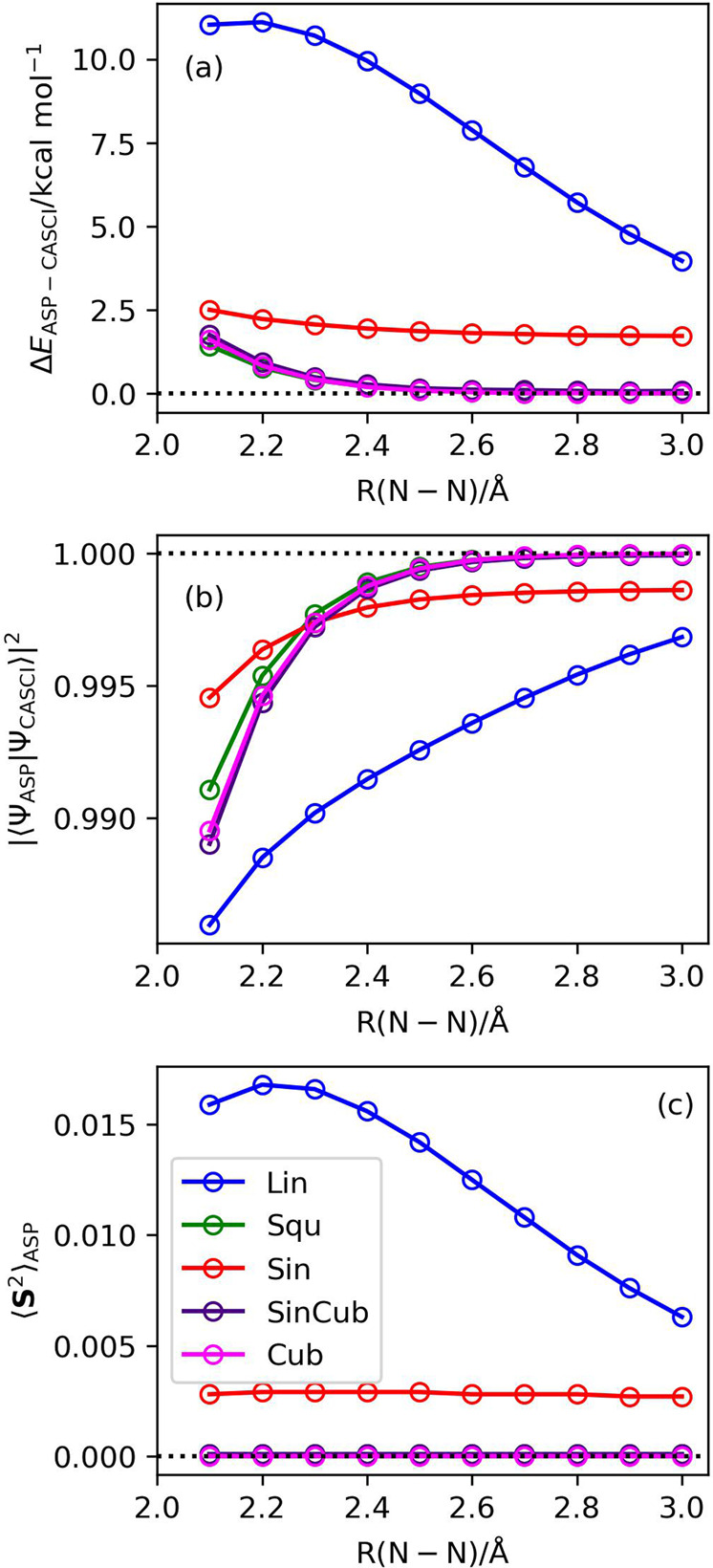
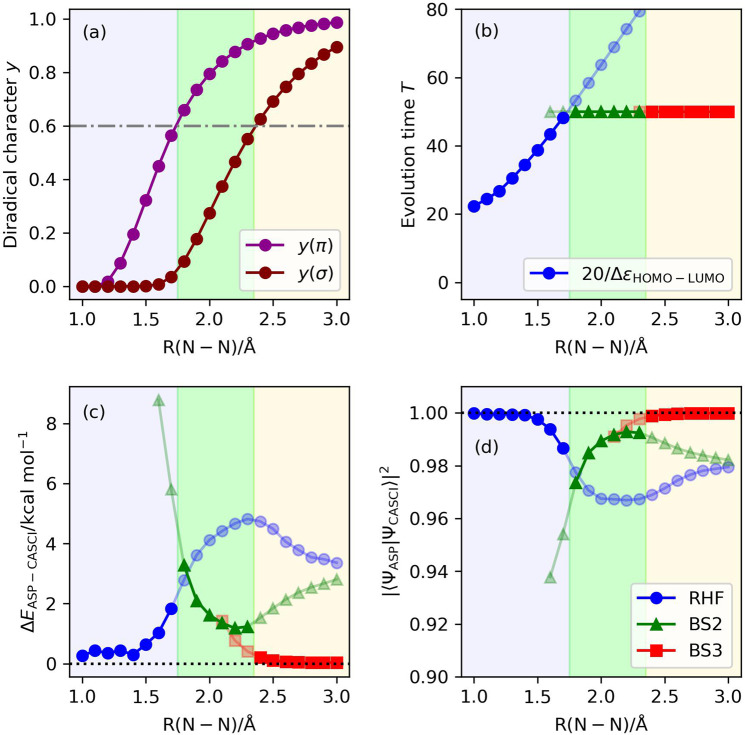
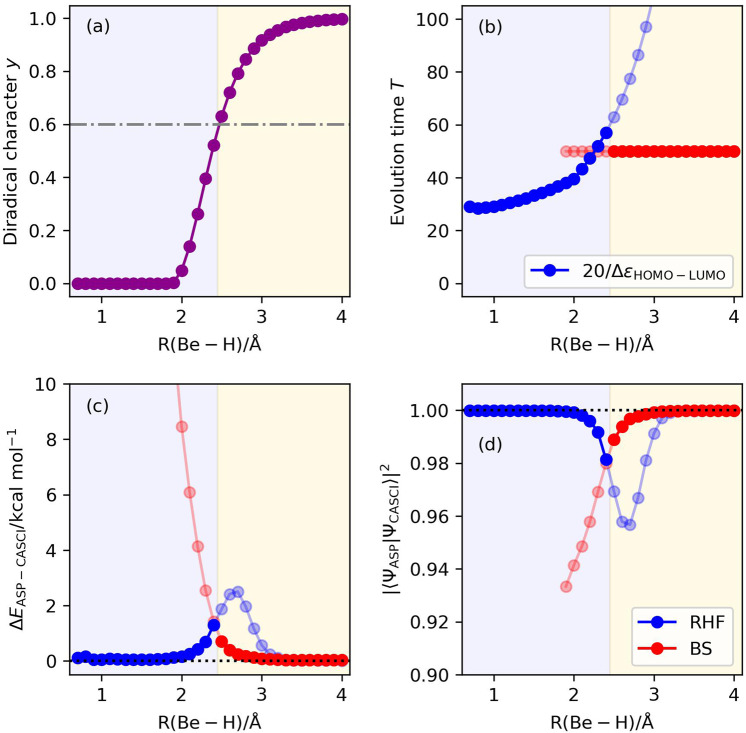
Adiabatic state preparation (ASP) can generate the correlated wave function by simulating the time evolution of wave function under the time-dependent Hamiltonian that interpolates the Fock operator and the full electronic Hamiltonian. However, ASP is inherently unsuitable for studying strongly correlated systems, and furthermore practical computational conditions for ASP are unknown. In quest for the suitable computational conditions for practical applications of ASP, we performed numerical simulations of ASP in the potential energy curves of N, BeH, and in the C quasi-reaction pathway of the Be atom insertion to the H molecule, examining the effect of nonlinear scheduling functions and the ASP with broken-symmetry wave functions with the S operator as the penalty term, contributing to practical applications of quantum computing to quantum chemistry. Eventually, computational guidelines to generate the correlated wave functions having the square overlap with the complete-active space self-consistent field wave function close to unity are discussed.
绝热态制备(ASP)可以通过在随时间变化的哈密顿量下模拟波函数的时间演化来生成相关波函数,该哈密顿量对福克算符和完整电子哈密顿量进行插值。然而,ASP本质上不适合研究强关联系统,而且ASP的实际计算条件尚不清楚。为了寻找ASP实际应用的合适计算条件,我们在N、BeH的势能曲线以及Be原子插入H分子的C准反应路径中对ASP进行了数值模拟,研究了非线性调度函数和以S算符为惩罚项的具有破缺对称性波函数的ASP的效果,这有助于量子计算在量子化学中的实际应用。最终,讨论了生成与完全活性空间自洽场波函数的平方重叠接近1的相关波函数的计算指南。