Sugisaki Kenji, Nakazawa Shigeaki, Toyota Kazuo, Sato Kazunobu, Shiomi Daisuke, Takui Takeji
Department of Chemistry and Molecular Materials Science, Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan.
Research Support Department/University Research Administrator Center, University Administration Division, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan.
ACS Cent Sci. 2019 Jan 23;5(1):167-175. doi: 10.1021/acscentsci.8b00788. Epub 2018 Dec 31.
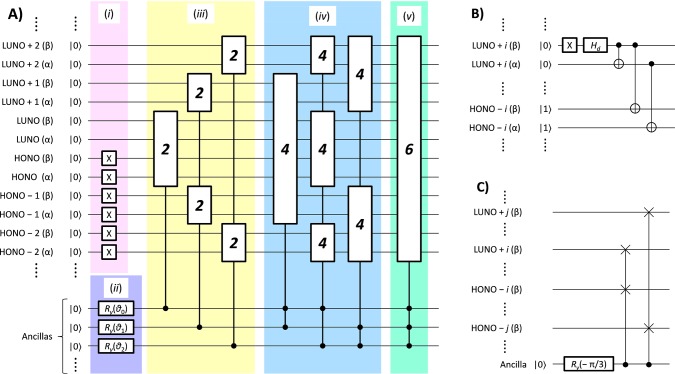
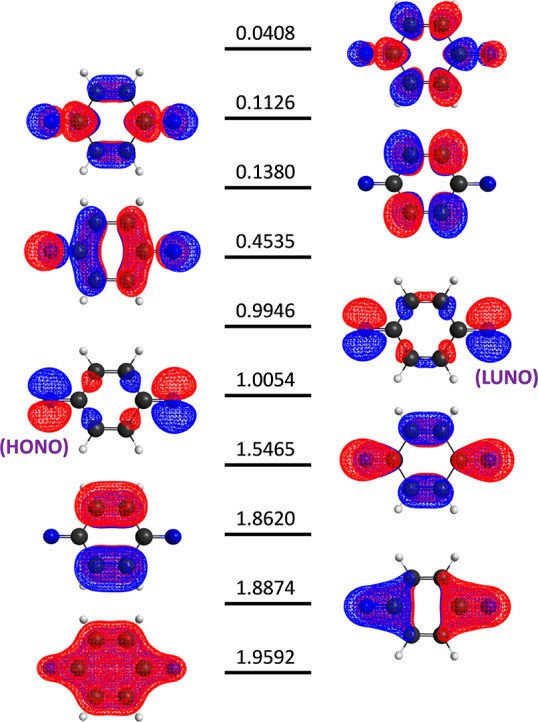
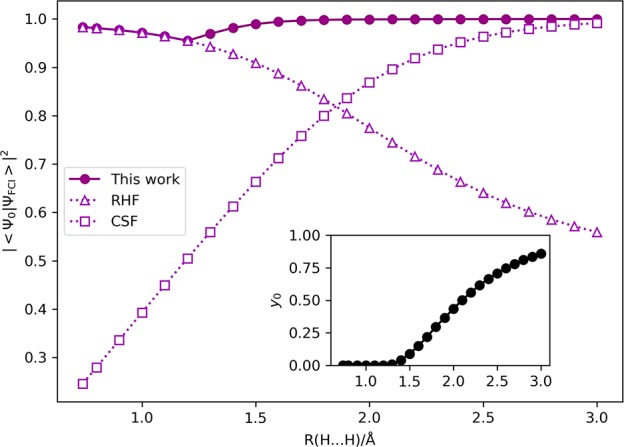
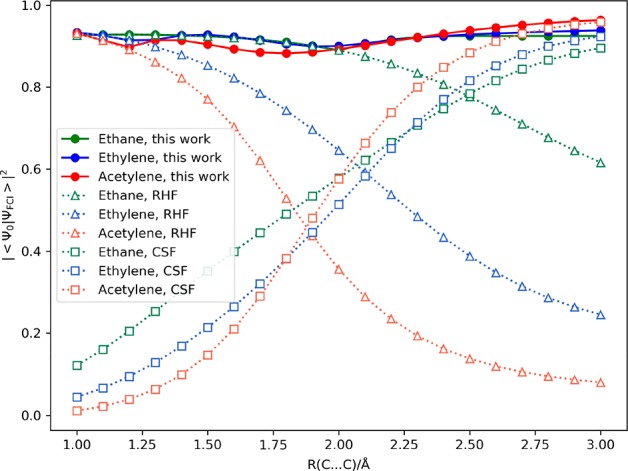
The full configuration interaction (full-CI) method is capable of providing the numerically best wave functions and energies of atoms and molecules within basis sets being used, although it is intractable for classical computers. Quantum computers can perform full-CI calculations in polynomial time against the system size by adopting a quantum phase estimation algorithm (QPEA). In the QPEA, the preparation of initial guess wave functions having sufficiently large overlap with the exact wave function is recommended. The Hartree-Fock (HF) wave function is a good initial guess only for closed shell singlet molecules and high-spin molecules carrying no spin-β unpaired electrons, around their equilibrium geometry, and thus, the construction of multiconfigurational wave functions without performing post-HF calculations on classical computers is highly desired for applying the method to a wide variety of chemistries and physics. In this work, we propose a method to construct multiconfigurational initial guess wave functions suitable for QPEA-based full-CI calculations on quantum computers, by utilizing diradical characters computed from spin-projected UHF wave functions. The proposed approach drastically improves the wave function overlap, particularly in molecules with intermediate diradical characters.
全组态相互作用(full-CI)方法能够在所用基组范围内提供原子和分子的数值上最优的波函数和能量,尽管它对于经典计算机来说是难以处理的。量子计算机可以通过采用量子相位估计算法(QPEA),在与系统大小相关的多项式时间内执行全CI计算。在QPEA中,建议制备与精确波函数有足够大重叠的初始猜测波函数。哈特里-福克(HF)波函数仅对于闭壳层单重态分子以及在其平衡几何构型附近不携带自旋-β未配对电子的高自旋分子是一个好的初始猜测,因此,对于将该方法应用于广泛的化学和物理领域而言,非常需要在不使用经典计算机进行后HF计算的情况下构建多组态波函数。在这项工作中,我们提出了一种方法,通过利用从自旋投影的UHF波函数计算出的双自由基特征,来构建适用于量子计算机上基于QPEA的全CI计算的多组态初始猜测波函数。所提出的方法极大地改善了波函数重叠,特别是在具有中等双自由基特征的分子中。