Department of Chemistry, Graduate School of Science, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka558-8585, Japan.
JSTPRESTO, 4-1-8 Honcho, Kawaguchi, Saitama, 332-0012, Japan.
J Phys Chem Lett. 2022 Dec 8;13(48):11105-11111. doi: 10.1021/acs.jpclett.2c02737. Epub 2022 Nov 29.
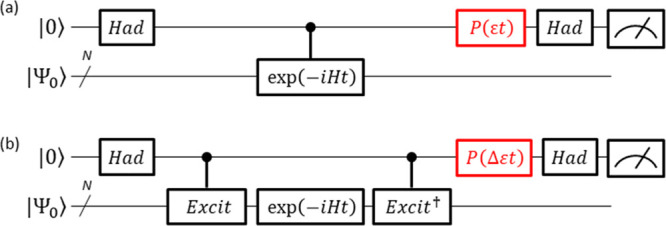
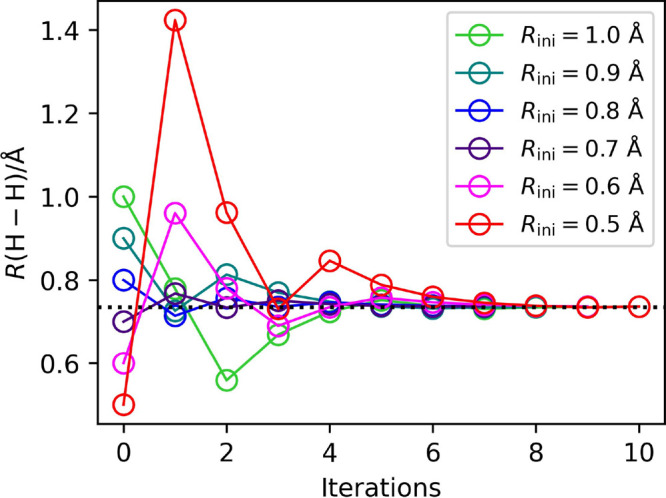
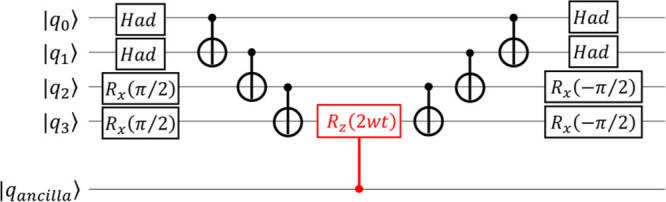
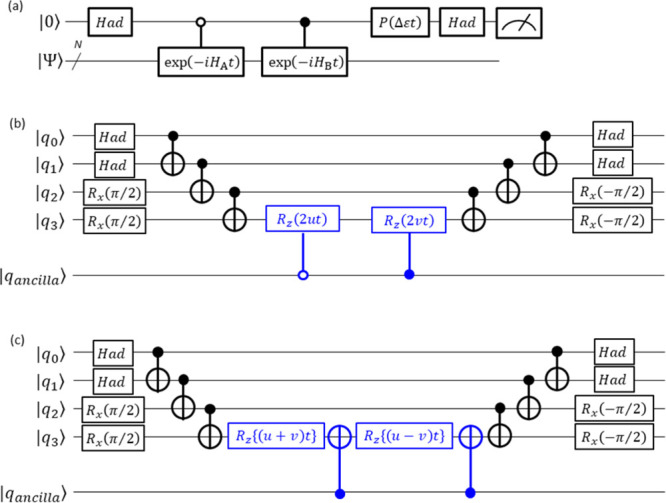
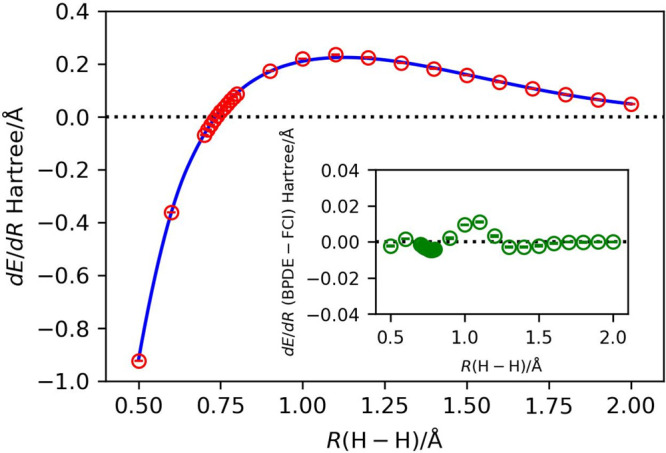
A Bayesian phase difference estimation (BPDE) algorithm allows us to compute the energy gap of two electronic states of a given Hamiltonian directly by utilizing the quantum superposition of their wave functions. Here we report an extension of the BPDE algorithm to the direct calculation of the energy difference of two molecular geometries. We apply the BPDE algorithm for the calculation of numerical energy gradients based on the two-point finite-difference method, enabling us to execute geometry optimization of one-dimensional molecules at the full-CI level on a quantum computer. Results of numerical quantum circuit simulations of the geometry optimization of the H molecule with the STO-3G and 6-31G basis sets, the LiH and BeH molecules at the full-CI/STO-3G level, and the N molecule at the CASCI(6e,6o)/6-311G* level are given.
贝叶斯相位差估计(BPDE)算法允许我们通过利用波函数的量子叠加,直接计算给定哈密顿量的两个电子态的能量差。在这里,我们报告了 BPDE 算法的扩展,用于直接计算两个分子构象的能量差。我们应用 BPDE 算法基于两点有限差分法计算数值能量梯度,使我们能够在量子计算机上执行全 CI 水平的一维分子的几何优化。给出了 STO-3G 和 6-31G 基组的 H 分子、全 CI/STO-3G 水平的 LiH 和 BeH 分子以及 CASCI(6e,6o)/6-311G*水平的 N 分子的几何优化的数值量子电路模拟结果。