Yang Hsin-Chou, Wang Jen-Hung, Yang Chih-Ting, Lin Yin-Chun, Hsieh Han-Ni, Chen Po-Wen, Liao Hsiao-Chi, Chen Chun-Houh, Liao James C
Institute of Statistical Science, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
Institute of Biological Chemistry, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
PNAS Nexus. 2022 Sep 1;1(4):pgac181. doi: 10.1093/pnasnexus/pgac181. eCollection 2022 Sep.
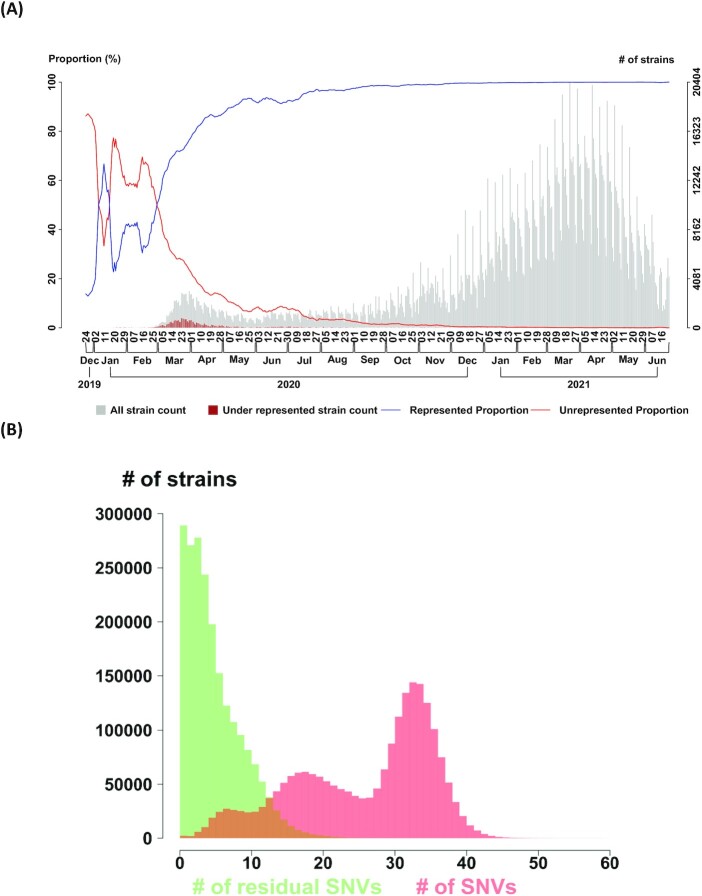
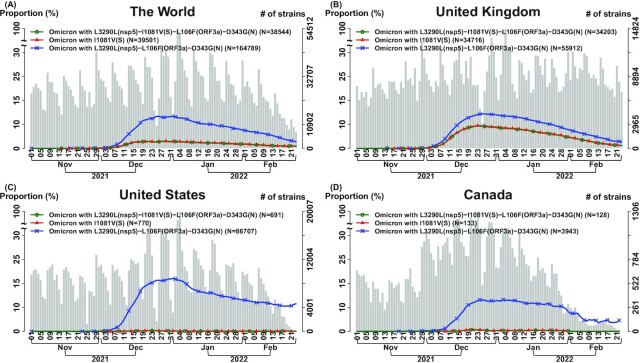
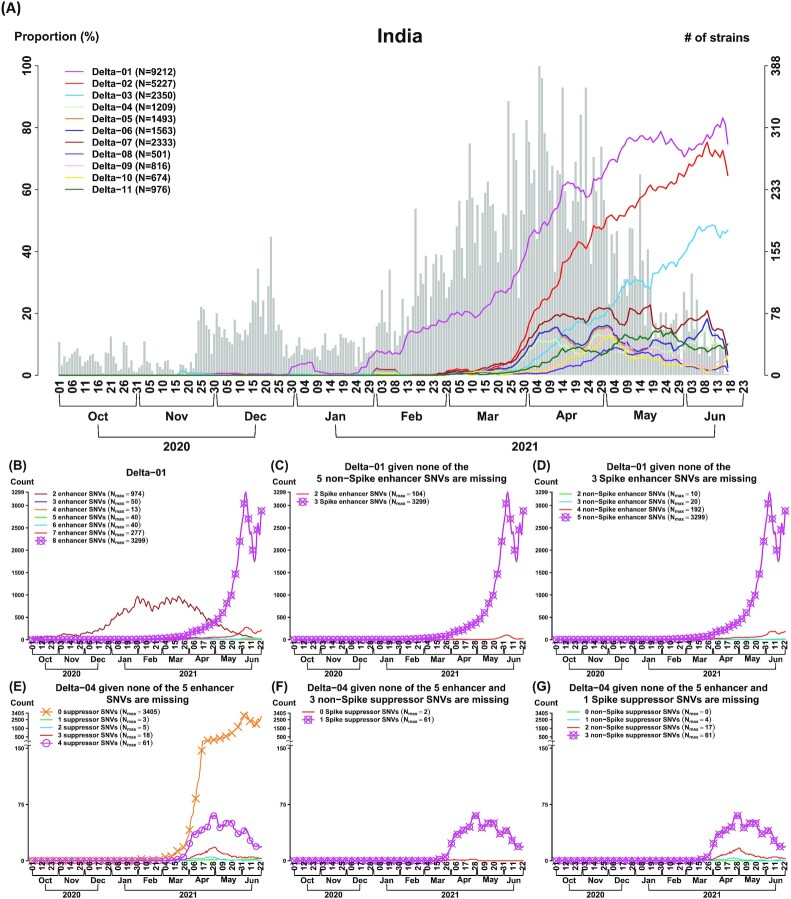
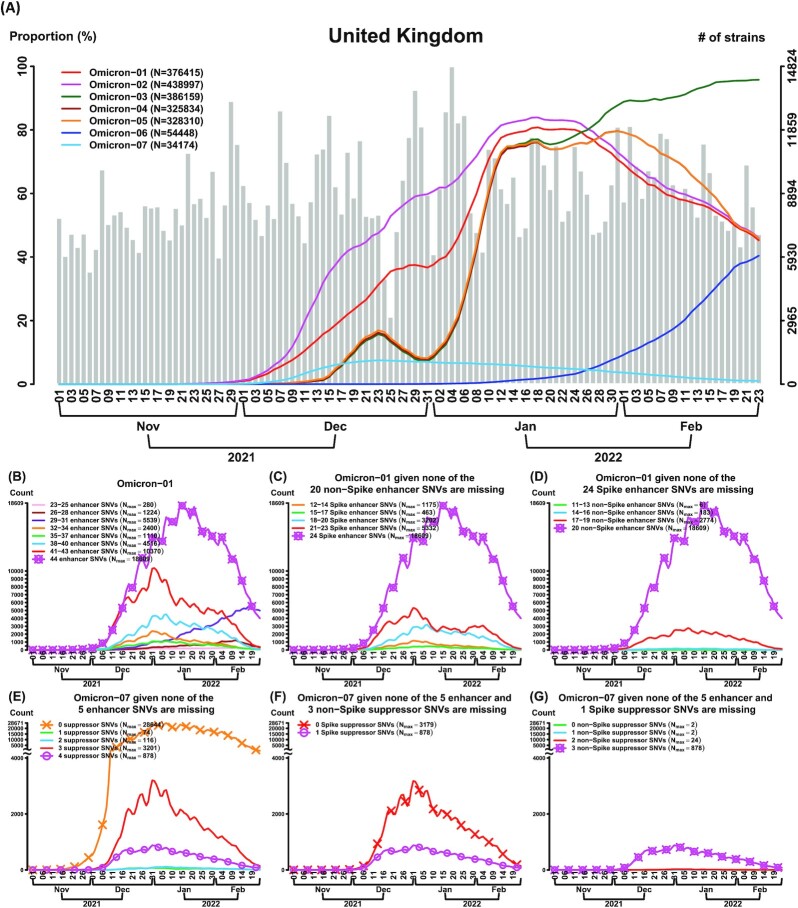
SARS-CoV-2 continues to evolve, causing waves of the pandemic. Up to May 2022, 10 million genome sequences have accumulated, which are classified into five major variants of concern. With the growing number of sequenced genomes, analysis of the big dataset has become increasingly challenging. Here we developed systematic approaches based on sets of correlated single nucleotide variations (SNVs) for comprehensive subtyping and pattern recognition of transmission dynamics. The approach outperformed single-SNV and spike-centric scans. Moreover, the derived subtypes elucidate the relationship of signature SNVs and transmission dynamics. We found that different subtypes of the same variant, including Delta and Omicron exhibited distinct temporal trajectories. For example, some Delta and Omicron subtypes did not spread rapidly, while others did. We identified sets of characteristic SNVs that appeared to enhance transmission or decrease efficacy of antibodies for some subtypes. We also identified a set of SNVs that appeared to suppress transmission or increase viral sensitivity to antibodies. For the Omicron variant, the dominant type in the world, we identified the subtypes with enhanced and suppressed transmission in an analysis of eight million genomes as of March 2022 and further confirmed the findings in a later analysis of ten million genomes as of May 2022. While the "enhancer" SNVs exhibited an enriched presence on the spike protein, the "suppressor" SNVs are mainly elsewhere. Disruption of the SNV correlation largely destroyed the enhancer-suppressor phenomena. These results suggest the importance of fine subtyping of variants, and point to potential complex interactions among SNVs.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)持续进化,引发多波疫情。截至2022年5月,已积累了1000万个基因组序列,这些序列被分为五个主要关注变体。随着测序基因组数量的不断增加,对这个大数据集的分析变得越来越具有挑战性。在此,我们基于相关单核苷酸变异(SNV)集开发了系统方法,用于对传播动态进行全面的亚型分类和模式识别。该方法优于单SNV和以刺突蛋白为中心的扫描。此外,所衍生的亚型阐明了特征性SNV与传播动态之间的关系。我们发现,同一变体的不同亚型,包括德尔塔和奥密克戎,表现出不同的时间轨迹。例如,一些德尔塔和奥密克戎亚型传播并不迅速,而其他亚型则不然。我们确定了一些特征性SNV集,这些SNV似乎增强了某些亚型的传播或降低了抗体的效力。我们还确定了一组似乎抑制传播或增加病毒对抗体敏感性的SNV。对于全球占主导地位的奥密克戎变体,我们在对截至2022年3月的800万个基因组进行分析时确定了传播增强和受抑制的亚型,并在后来对截至2022年5月的1000万个基因组进行分析时进一步证实了这些发现。虽然“增强子”SNV在刺突蛋白上富集存在,但“抑制子”SNV主要在其他位置。SNV相关性的破坏在很大程度上消除了增强子-抑制子现象。这些结果表明对变体进行精细亚型分类的重要性,并指出了SNV之间潜在的复杂相互作用。