Morimoto Marie, Bhambhani Vikas, Gazzaz Nour, Davids Mariska, Sathiyaseelan Paalini, Macnamara Ellen F, Lange Jennifer, Lehman Anna, Zerfas Patricia M, Murphy Jennifer L, Acosta Maria T, Wang Camille, Alderman Emily, Reichert Sara, Thurm Audrey, Adams David R, Introne Wendy J, Gorski Sharon M, Boerkoel Cornelius F, Gahl William A, Tifft Cynthia J, Malicdan May Christine V
National Institutes of Health Undiagnosed Diseases Program, Common Fund, Office of the Director, National Institutes of Health, Bethesda, MD, 20892, USA.
Department of Medical Genetics, Children's Hospitals and Clinics of Minnesota, Minneapolis, MN, 55404, USA.
NPJ Genom Med. 2023 Feb 10;8(1):4. doi: 10.1038/s41525-022-00343-8.
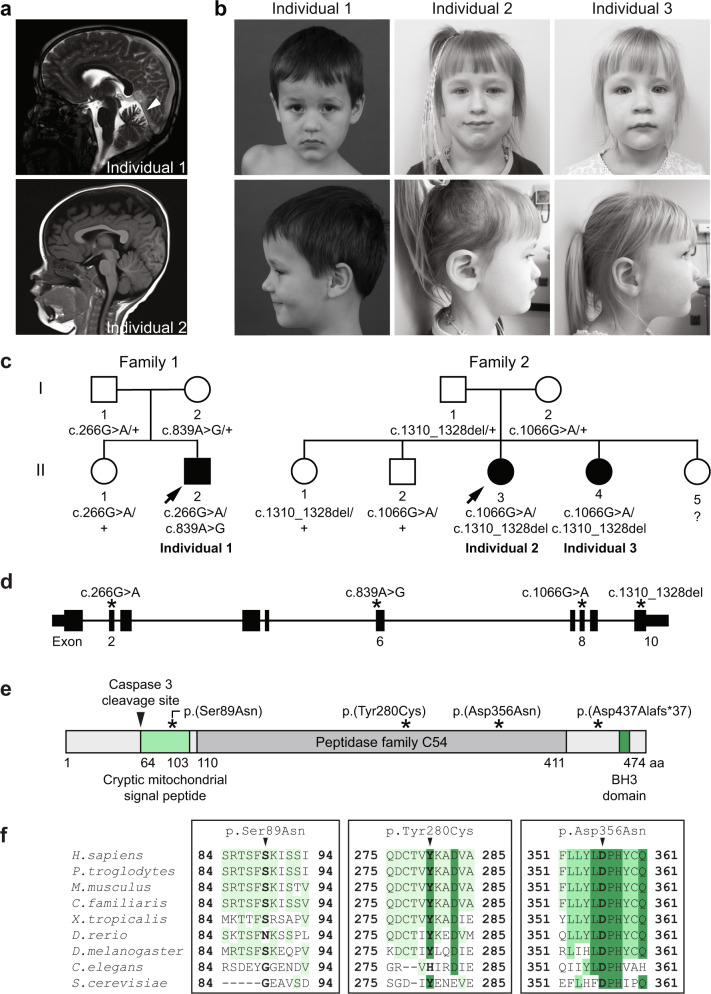
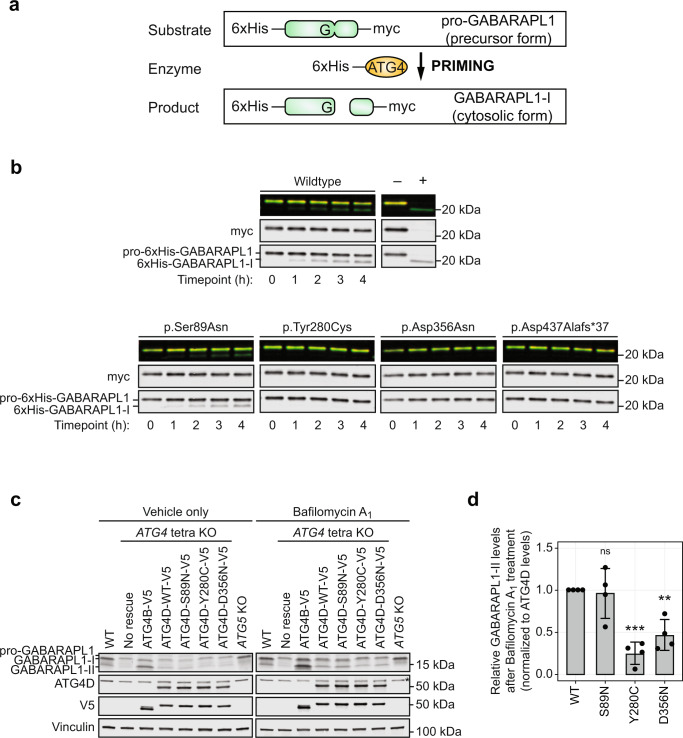
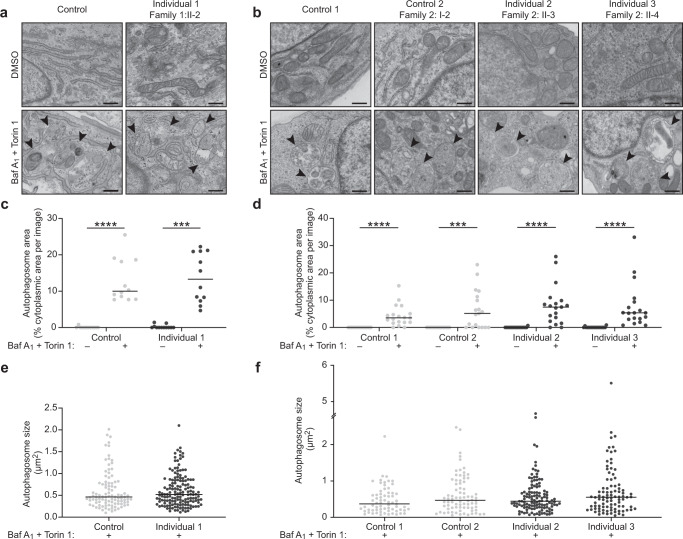
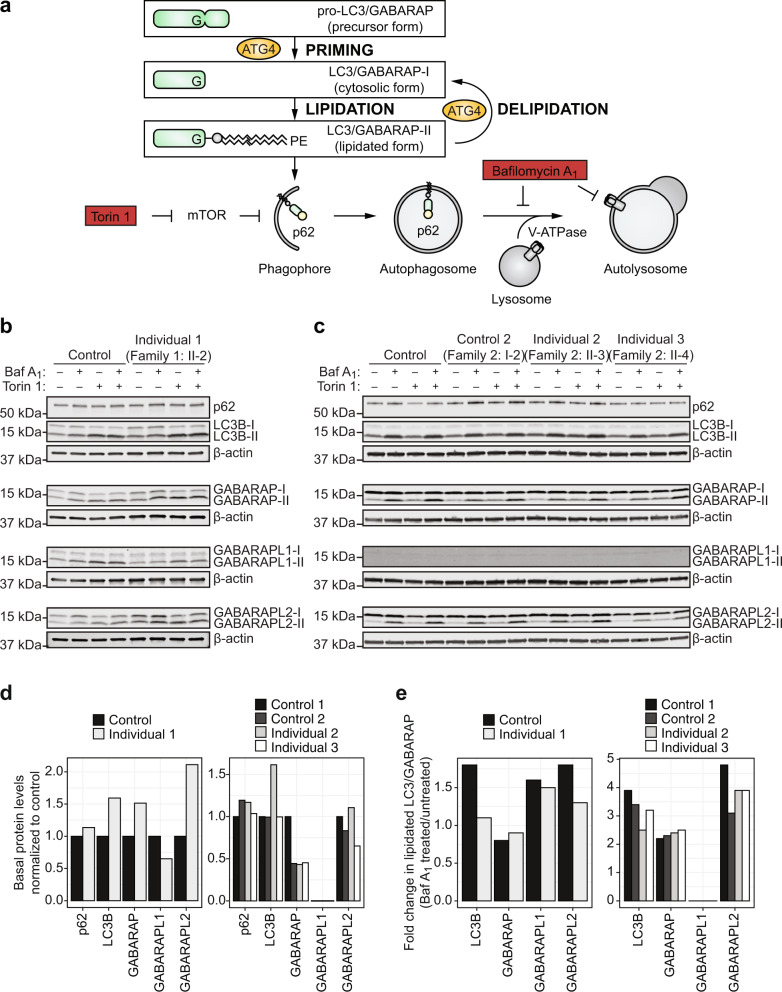
Autophagy regulates the degradation of damaged organelles and protein aggregates, and is critical for neuronal development, homeostasis, and maintenance, yet few neurodevelopmental disorders have been associated with pathogenic variants in genes encoding autophagy-related proteins. We report three individuals from two unrelated families with a neurodevelopmental disorder characterized by speech and motor impairment, and similar facial characteristics. Rare, conserved, bi-allelic variants were identified in ATG4D, encoding one of four ATG4 cysteine proteases important for autophagosome biogenesis, a hallmark of autophagy. Autophagosome biogenesis and induction of autophagy were intact in cells from affected individuals. However, studies evaluating the predominant substrate of ATG4D, GABARAPL1, demonstrated that three of the four ATG4D patient variants functionally impair ATG4D activity. GABARAPL1 is cleaved or "primed" by ATG4D and an in vitro GABARAPL1 priming assay revealed decreased priming activity for three of the four ATG4D variants. Furthermore, a rescue experiment performed in an ATG4 tetra knockout cell line, in which all four ATG4 isoforms were knocked out by gene editing, showed decreased GABARAPL1 priming activity for the two ATG4D missense variants located in the cysteine protease domain required for priming, suggesting that these variants impair the function of ATG4D. The clinical, bioinformatic, and functional data suggest that bi-allelic loss-of-function variants in ATG4D contribute to the pathogenesis of this syndromic neurodevelopmental disorder.
自噬调节受损细胞器和蛋白质聚集体的降解,对神经元发育、稳态和维持至关重要,但很少有神经发育障碍与编码自噬相关蛋白的基因中的致病变异相关。我们报告了来自两个无关家庭的三名个体,他们患有以言语和运动障碍以及相似面部特征为特征的神经发育障碍。在ATG4D中鉴定出罕见的、保守的双等位基因变体,该基因编码对自噬体生物发生(自噬的一个标志)很重要的四种ATG4半胱氨酸蛋白酶之一。来自受影响个体的细胞中的自噬体生物发生和自噬诱导是完整的。然而,评估ATG4D主要底物GABARAPL1的研究表明,四个ATG4D患者变体中的三个在功能上损害了ATG4D活性。GABARAPL1被ATG4D切割或“启动”,体外GABARAPL1启动试验显示四个ATG4D变体中的三个的启动活性降低。此外,在一个ATG4四敲除细胞系中进行的拯救实验表明,位于启动所需的半胱氨酸蛋白酶结构域中的两个ATG4D错义变体的GABARAPL1启动活性降低,这表明这些变体损害了ATG4D的功能。临床、生物信息学和功能数据表明,ATG4D中的双等位基因功能丧失变体促成了这种综合征性神经发育障碍的发病机制。