Department of Chemistry, Stanford University, Stanford, CA, USA.
Department of Computer Science, Stanford University, Stanford, CA, USA.
Nat Chem Biol. 2023 Jul;19(7):805-814. doi: 10.1038/s41589-022-01247-5. Epub 2023 Feb 13.
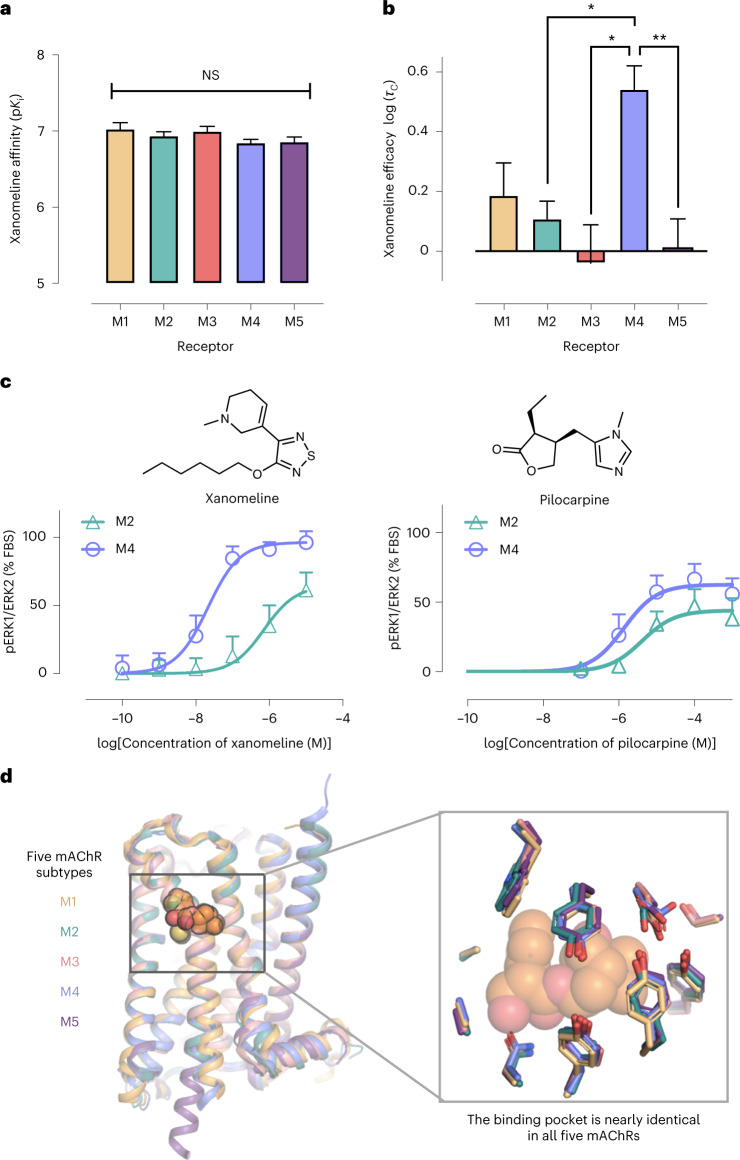
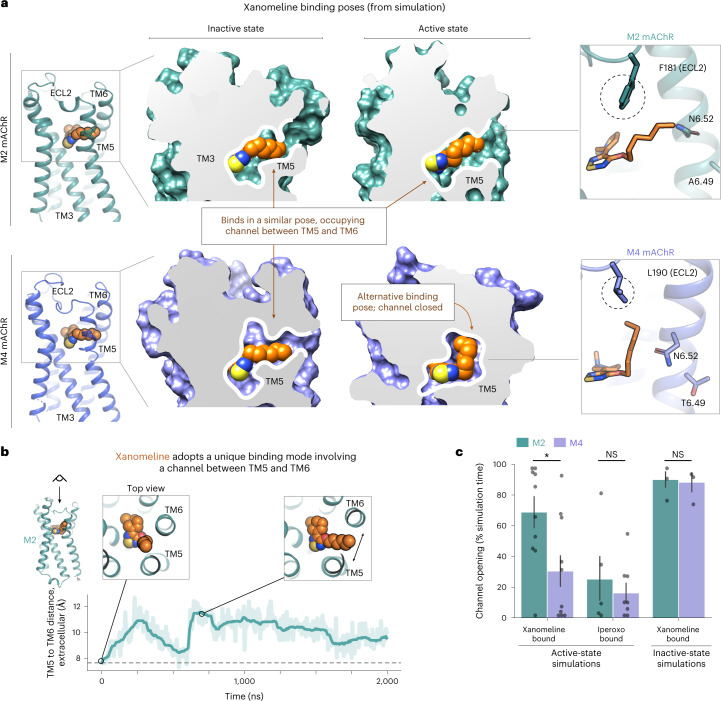
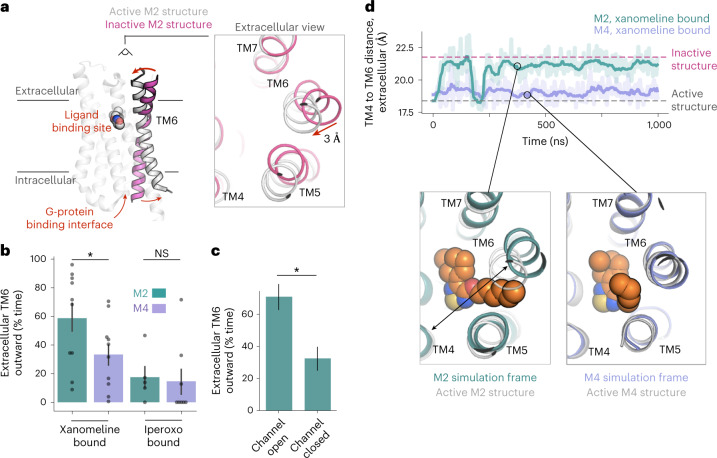
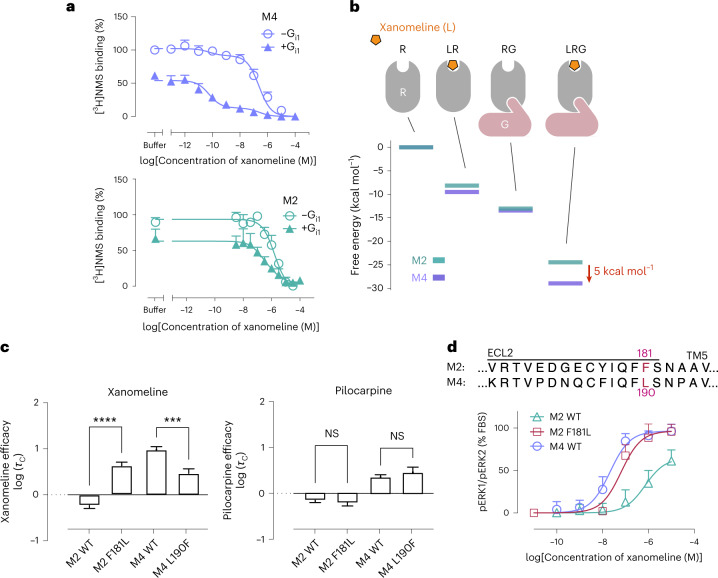
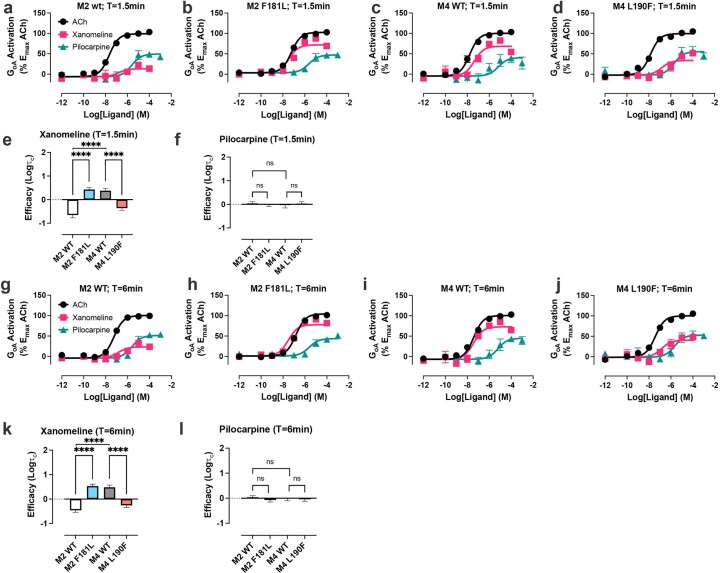
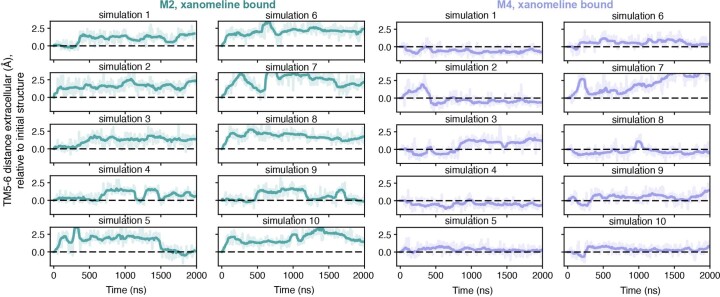
A drug's selectivity for target receptors is essential to its therapeutic utility, but achieving selectivity between similar receptors is challenging. The serendipitous discovery of ligands that stimulate target receptors more strongly than closely related receptors, despite binding with similar affinities, suggests a solution. The molecular mechanism of such 'efficacy-driven selectivity' has remained unclear, however, hindering design of such ligands. Here, using atomic-level simulations, we reveal the structural basis for the efficacy-driven selectivity of a long-studied clinical drug candidate, xanomeline, between closely related muscarinic acetylcholine receptors (mAChRs). Xanomeline's binding mode is similar across mAChRs in their inactive states but differs between mAChRs in their active states, with divergent effects on active-state stability. We validate this mechanism experimentally and use it to design ligands with altered efficacy-driven selectivity. Our results suggest strategies for the rational design of ligands that achieve efficacy-driven selectivity for many pharmaceutically important G-protein-coupled receptors.
药物对靶受体的选择性对于其治疗用途至关重要,但在相似受体之间实现选择性具有挑战性。尽管具有相似的亲和力,但偶然发现的刺激靶受体的配体比密切相关的受体更强,这为解决这个问题提供了思路。然而,这种“效能驱动选择性”的分子机制仍不清楚,这阻碍了此类配体的设计。在这里,我们使用原子水平的模拟,揭示了一种长期研究的临床候选药物 xanomeline 在其密切相关的毒蕈碱型乙酰胆碱受体 (mAChR) 之间产生效能驱动选择性的结构基础。 xanomeline 在其非活性状态下与 mAChR 的结合模式相似,但在其活性状态下与 mAChR 不同,对活性状态稳定性有不同的影响。我们通过实验验证了这一机制,并利用它设计了具有改变的效能驱动选择性的配体。我们的结果表明了针对许多具有重要药物应用的 G 蛋白偶联受体实现效能驱动选择性的配体的合理设计策略。