Stergachis Andrew B, Blue Elizabeth E, Gillentine Madelyn A, Wang Lee-Kai, Schwarze Ulrike, Cortés Adriana Sedeño, Ranchalis Jane, Allworth Aimee, Bland Austin E, Chanprasert Sirisak, Chen Jingheng, Doherty Daniel, Folta Andrew B, Glass Ian, Horike-Pyne Martha, Huang Alden Y, Khan Alyna T, Leppig Kathleen A, Miller Danny E, Mirzaa Ghayda, Parhin Azma, Raskind Wendy, Rosenthal Elisabeth A, Sheppeard Sam, Strohbehn Samuel, Sybert Virginia P, Tran Thao T, Wener Mark, Byers Peter H, Nelson Stanley F, Bamshad Michael J, Dipple Katrina M, Jarvik Gail P, Hoppins Suzanne, Hisama Fuki M
University of Washington School of Medicine, Department of Medicine, Seattle, WA, USA.
University of Washington School of Medicine, Genome Sciences, Seattle, WA, USA.
bioRxiv. 2023 Feb 7:2023.02.07.526487. doi: 10.1101/2023.02.07.526487.
Transcript sequencing of patient derived samples has been shown to improve the diagnostic yield for solving cases of likely Mendelian disorders, yet the added benefit of full-length long-read transcript sequencing is largely unexplored.
We applied short-read and full-length isoform cDNA sequencing and mitochondrial functional studies to a patient-derived fibroblast cell line from an individual with neuropathy that previously lacked a molecular diagnosis.
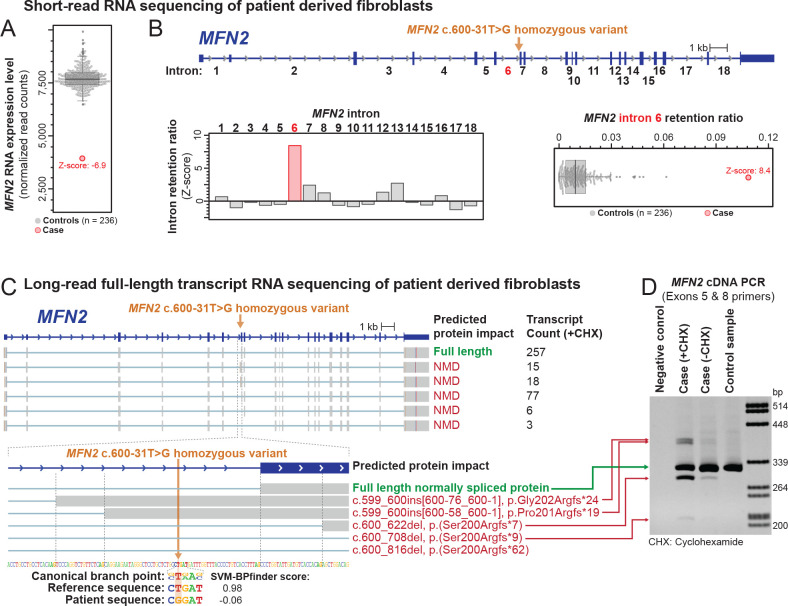
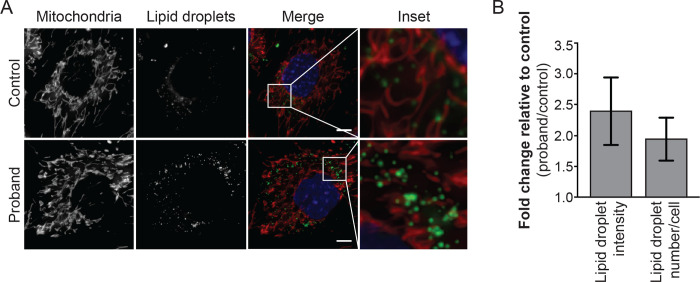
We identified an intronic homozygous c.600-31T>G variant that disrupts a branch point critical for intron 6 spicing. Full-length long-read isoform cDNA sequencing after treatment with a nonsense-mediated mRNA decay (NMD) inhibitor revealed that this variant creates five distinct altered splicing transcripts. All five altered splicing transcripts have disrupted open reading frames and are subject to NMD. Furthermore, a patient-derived fibroblast line demonstrated abnormal lipid droplet formation, consistent with MFN2 dysfunction. Although correctly spliced full-length transcripts are still produced, this branch point variant results in deficient MFN2 protein levels and autosomal recessive Charcot-Marie-Tooth disease, axonal, type 2A (CMT2A).
This case highlights the utility of full-length isoform sequencing for characterizing the molecular mechanism of undiagnosed rare diseases and expands our understanding of the genetic basis for CMT2A.
对患者来源的样本进行转录本测序已被证明可提高解决可能的孟德尔疾病病例的诊断率,但全长长读长转录本测序的额外益处很大程度上尚未得到探索。
我们对一名先前缺乏分子诊断的患有神经病变的个体的患者来源的成纤维细胞系应用了短读长和全长异构体cDNA测序以及线粒体功能研究。
我们鉴定出一个内含子纯合的c.600-31T>G变异,该变异破坏了对第6号内含子剪接至关重要的一个分支点。在用无义介导的mRNA衰变(NMD)抑制剂处理后进行的全长长读长异构体cDNA测序显示,该变异产生了五种不同的改变剪接的转录本。所有五种改变剪接的转录本都有破坏的开放阅读框,并受到NMD的影响。此外,患者来源的成纤维细胞系表现出异常的脂滴形成,这与MFN2功能障碍一致。尽管仍会产生正确剪接的全长转录本,但这个分支点变异导致MFN2蛋白水平不足,并引发常染色体隐性遗传性腓骨肌萎缩症2A型(CMT2A)。
该病例突出了全长异构体测序在表征未确诊罕见疾病分子机制方面的效用,并扩展了我们对CMT2A遗传基础的理解。