Department of Pediatrics, Dr. von Hauner Children's Hospital, University Hospital, Ludwig-Maximilians-University, Munich, Germany.
German Center for Lung Research (DZL), Munich, Germany.
Thorax. 2023 Jun;78(6):587-595. doi: 10.1136/thorax-2022-219434. Epub 2023 Feb 20.
The majority of patients with childhood interstitial lung disease (chILD) caused by pathogenic variants in ATP binding cassette subfamily A member 3 (ABCA3) develop severe respiratory insufficiency within their first year of life and succumb to disease if not lung transplanted. This register-based cohort study reviews patients with ABCA3 lung disease who survived beyond the age of 1 year.
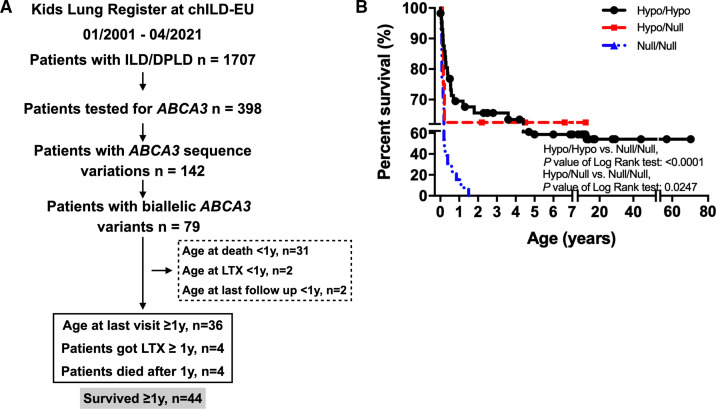
Over a 21-year period, patients diagnosed as chILD due to ABCA3 deficiency were identified from the Kids Lung Register database. 44 patients survived beyond the first year of life and their long-term clinical course, oxygen supplementation and pulmonary function were reviewed. Chest CT and histopathology were scored blindly.
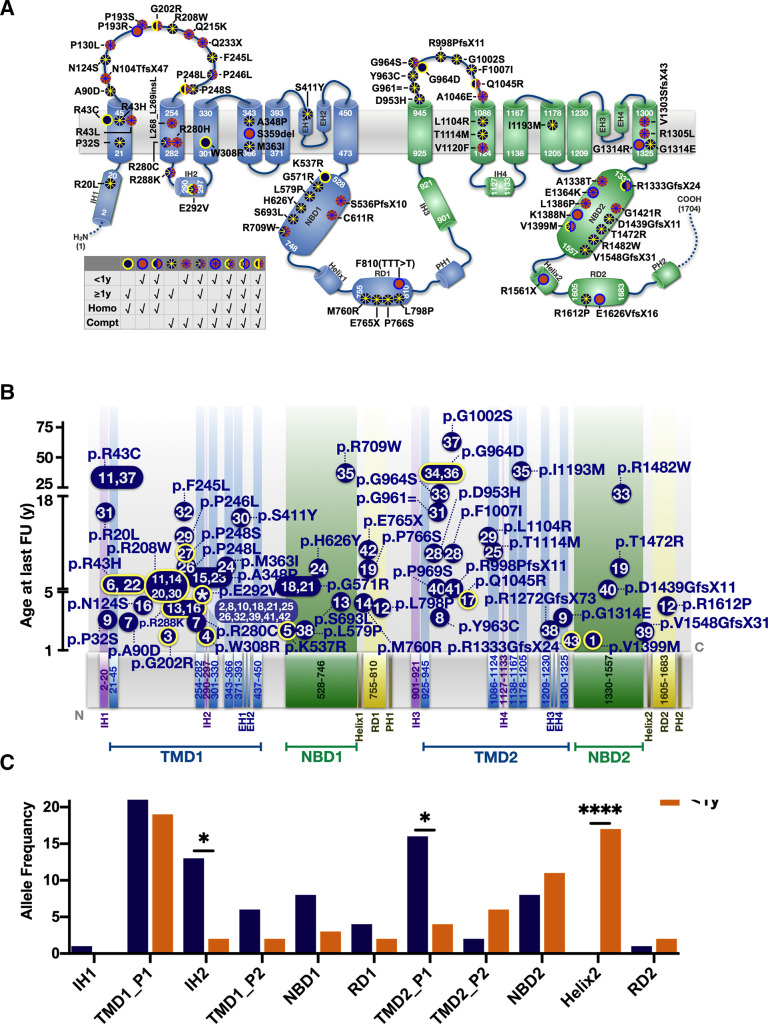
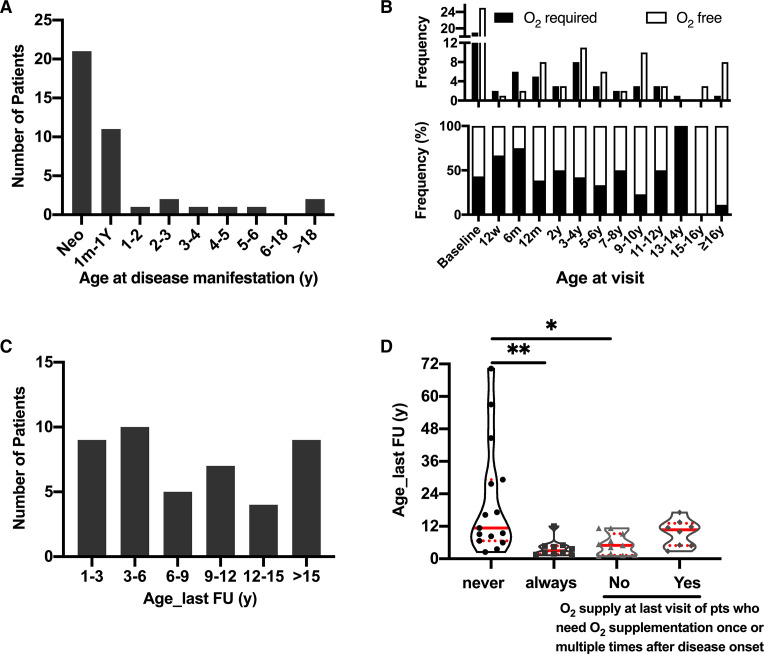
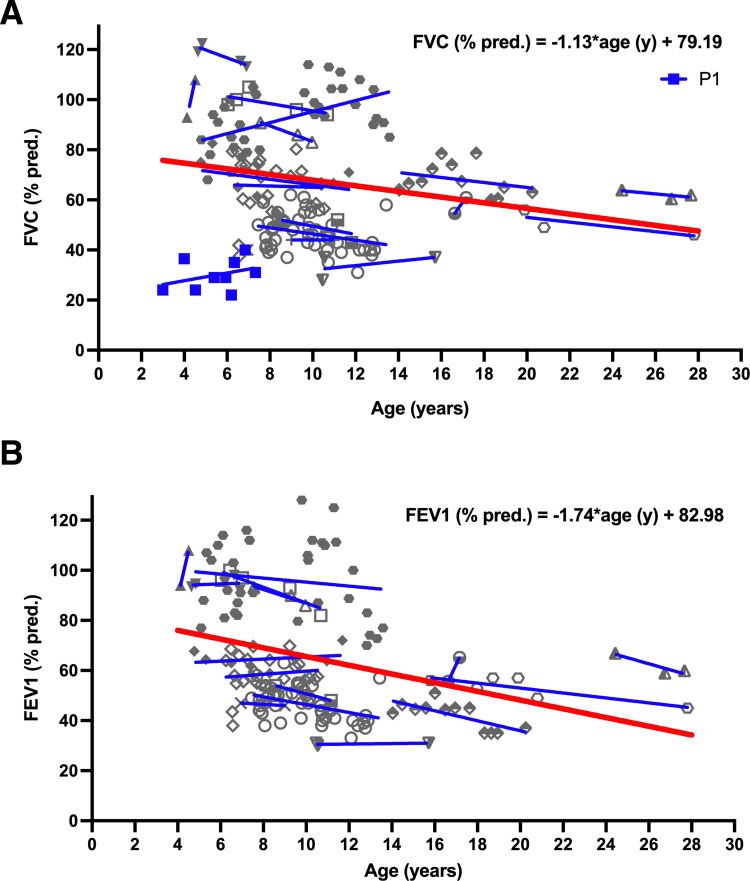
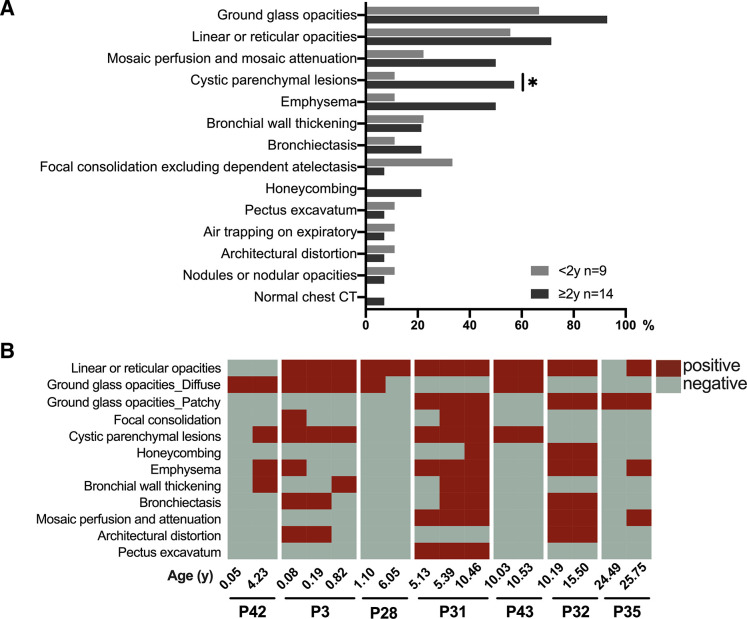
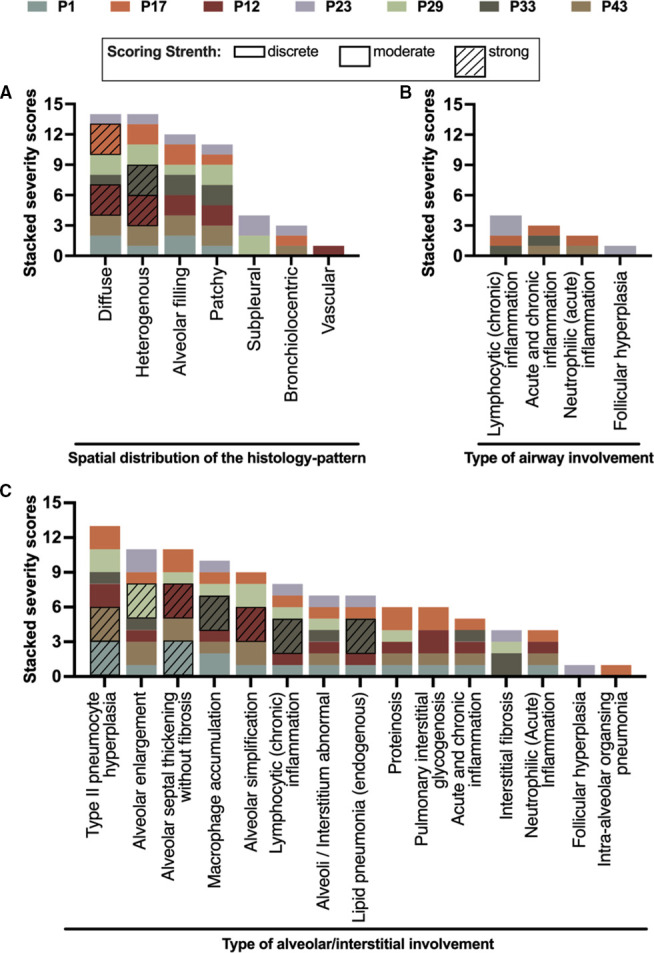
At the end of the observation period, median age was 6.3 years (IQR: 2.8-11.7) and 36/44 (82%) were still alive without transplantation. Patients who had never received supplemental oxygen therapy survived longer than those persistently required oxygen supplementation (9.7 (95% CI 6.7 to 27.7) vs 3.0 years (95% CI 1.5 to 5.0), p0.0126). Interstitial lung disease was clearly progressive over time based on lung function (forced vital capacity % predicted absolute loss -1.1% /year) and on chest CT (increasing cystic lesions in those with repetitive imaging). Lung histology pattern were variable (chronic pneumonitis of infancy, non-specific interstitial pneumonia, and desquamative interstitial pneumonia). In 37/44 subjects, the sequence variants were missense variants, small insertions or deletions with in-silico tools predicting some residual ABCA3 transporter function.
The natural history of ABCA3-related interstitial lung disease progresses during childhood and adolescence. Disease-modifying treatments are desirable to delay such disease course.
大多数由 ATP 结合盒亚家族 A 成员 3(ABCA3)致病性变异引起的儿童间质性肺病(chILD)患者在生命的第一年就会出现严重的呼吸功能不全,如果不进行肺移植,就会死于该病。本基于注册的队列研究回顾了 ABCA3 肺疾病患者,这些患者的存活年龄超过了 1 岁。
在 21 年的时间里,从儿童肺登记数据库中确定了因 ABCA3 缺乏而被诊断为 chILD 的患者。44 名患者的存活年龄超过了 1 岁,对他们的长期临床过程、氧疗和肺功能进行了回顾。对胸部 CT 和组织病理学进行了盲法评分。
在观察期末,中位年龄为 6.3 岁(IQR:2.8-11.7),36/44(82%)仍未接受移植而存活。从未接受过氧疗的患者比持续需要氧疗的患者存活时间更长(9.7(95%CI 6.7 至 27.7)vs 3.0 年(95%CI 1.5 至 5.0),p=0.0126)。基于肺功能(用力肺活量%预测绝对值损失-1.1%/年)和胸部 CT(重复成像时出现更多囊性病变),间质性肺病随时间推移呈明显进展。肺组织病理学模式多样(婴儿期慢性肺炎、非特异性间质性肺炎和脱屑性间质性肺炎)。在 44 名患者中,序列变异为错义变异、小插入或缺失,预测一些残余的 ABCA3 转运体功能。
ABCA3 相关间质性肺疾病的自然病史在儿童和青少年时期进展。需要进行疾病修饰治疗以延缓疾病进程。