Ramos Daniel R, Furtmüller Paul G, Obinger Christian, Peña-Gallego Ángeles, Pérez-Juste Ignacio, Santaballa J Arturo
Chemical Reactivity & Photoreactivity Group (React!), Department of Chemistry, CICA & Faculty of Sciences, Universidade da Coruña, Campus da Zapateira, E-15071 A Coruña, Spain.
Departamento de Química Física, Universidade de Vigo, Campus Universitario Lagoas-Marcosende, E-36310 Vigo, Spain.
Antioxidants (Basel). 2023 Jan 28;12(2):303. doi: 10.3390/antiox12020303.
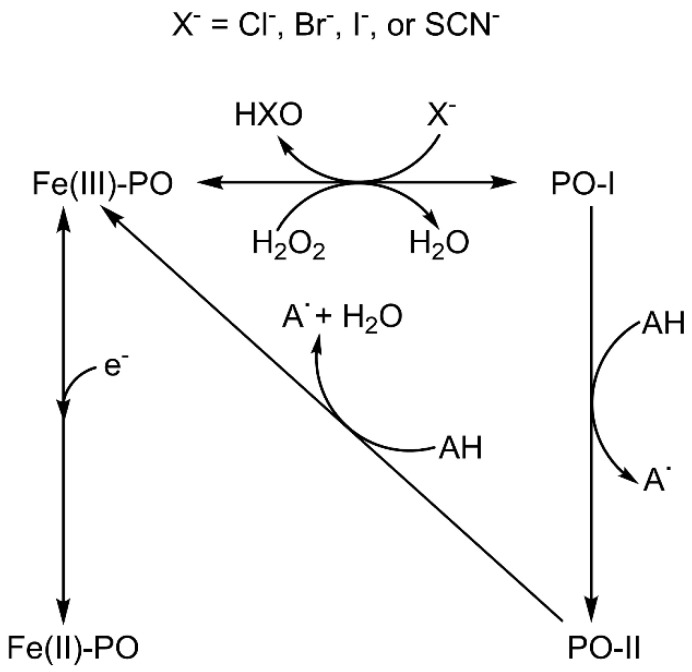
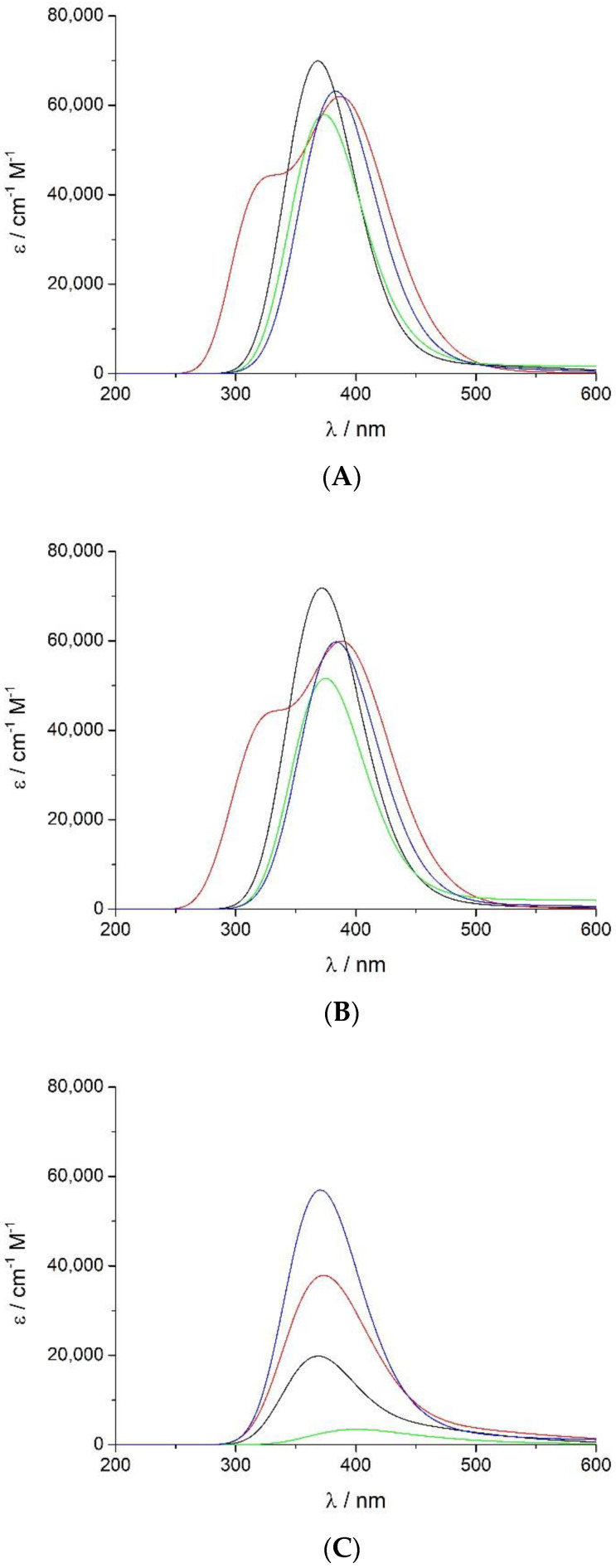
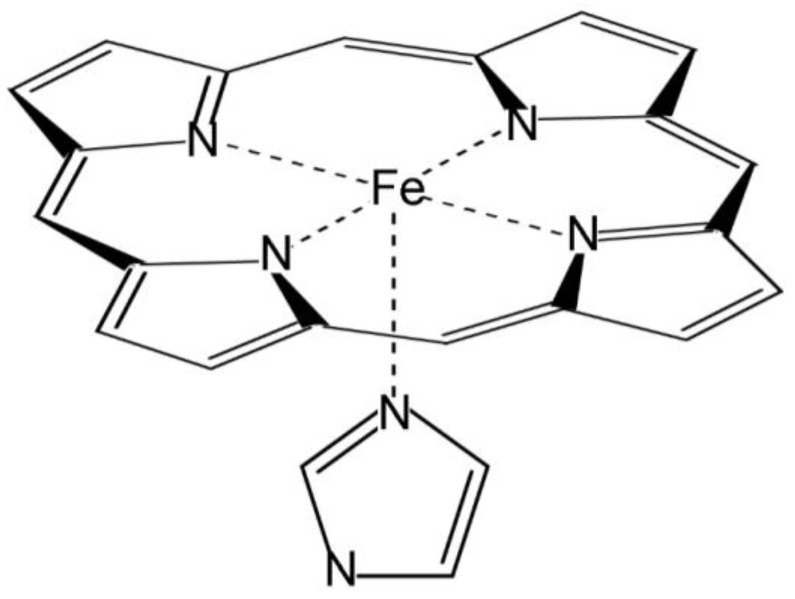
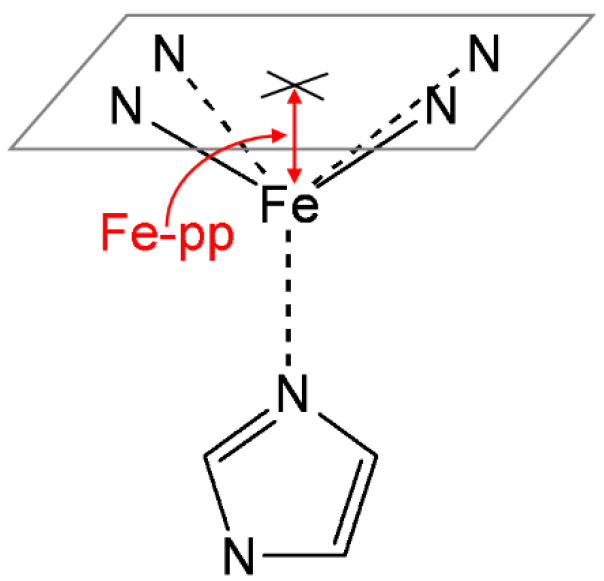
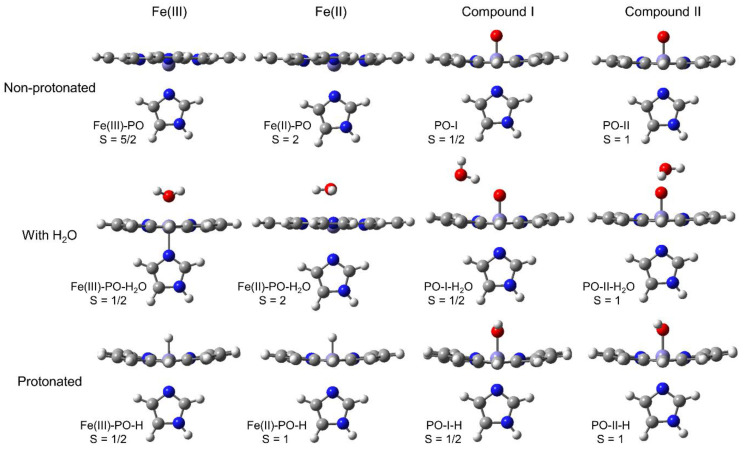
Electronic structure calculations using the density-functional theory (DFT) have been performed to analyse the effect of water molecules and protonation on the heme group of peroxidases in different redox (ferric, ferrous, compounds I and II) and spin states. Shared geometries, spectroscopic properties at the Soret region, and the thermodynamics of peroxidases are discussed. B3LYP and M06-2X density functionals with different basis sets were employed on a common molecular model of the active site (Fe-centred porphine and proximal imidazole). Computed Gibbs free energies indicate that the corresponding aquo complexes are not thermodynamically stable, supporting the five-coordinate Fe(III) centre in native ferric peroxidases, with a water molecule located at a non-bonding distance. Protonation of the ferryl oxygen of compound II is discussed in terms of thermodynamics, Fe-O bond distances, and redox properties. It is demonstrated that this protonation is necessary to account for the experimental data, and computed Gibbs free energies reveal p values of compound II about 8.5-9.0. Computation indicates that the general oxidative properties of peroxidase intermediates, as well as their reactivity towards water and protons and Soret bands, are mainly controlled by the iron porphyrin and its proximal histidine ligand.
已使用密度泛函理论(DFT)进行电子结构计算,以分析水分子和质子化对处于不同氧化还原状态(铁离子、亚铁离子、化合物I和II)及自旋态的过氧化物酶血红素基团的影响。讨论了过氧化物酶的共享几何结构、Soret区域的光谱性质以及热力学性质。在活性位点(以铁为中心的卟啉和近端咪唑)的通用分子模型上,采用了具有不同基组的B3LYP和M06 - 2X密度泛函。计算得到的吉布斯自由能表明相应的水合配合物在热力学上不稳定,这支持了天然铁离子过氧化物酶中五配位的Fe(III)中心,其中一个水分子位于非键合距离处。从热力学、Fe - O键距离和氧化还原性质等方面讨论了化合物II中高铁氧的质子化。结果表明,这种质子化对于解释实验数据是必要的,计算得到的吉布斯自由能显示化合物II的p值约为8.5 - 9.0。计算表明,过氧化物酶中间体的一般氧化性质,以及它们对水、质子和Soret带的反应性,主要由铁卟啉及其近端组氨酸配体控制。