Ibrahim Amy, Manko Emilia, Dombrowski Jamille G, Campos Mónica, Benavente Ernest Diez, Nolder Debbie, Sutherland Colin J, Nosten Francois, Fernandez Diana, Vélez-Tobón Gabriel, Castaño Alberto Tobón, Aguiar Anna Caroline C, Pereira Dhelio Batista, da Silva Santos Simone, Suarez-Mutis Martha, Di Santi Silvia Maria, Regina de Souza Baptista Andrea, Dantas Machado Ricardo Luiz, Marinho Claudio R F, Clark Taane G, Campino Susana
Faculty of Infectious & Tropical Diseases, London School of Hygiene & Tropical Medicine, London, UK.
Department of Parasitology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil.
Lancet Reg Health Am. 2023 Jan 2;18:100420. doi: 10.1016/j.lana.2022.100420. eCollection 2023 Feb.
Brazil is a unique and understudied setting for malaria, with complex foci of transmission associated with human and environmental conditions. An understanding of the population genomic diversity of parasites across Brazil can support malaria control strategies.
Through whole genome sequencing of isolates across 7 Brazilian states, we use population genomic approaches to compare genetic diversity within country (n = 123), continent (6 countries, n = 315) and globally (26 countries, n = 885).
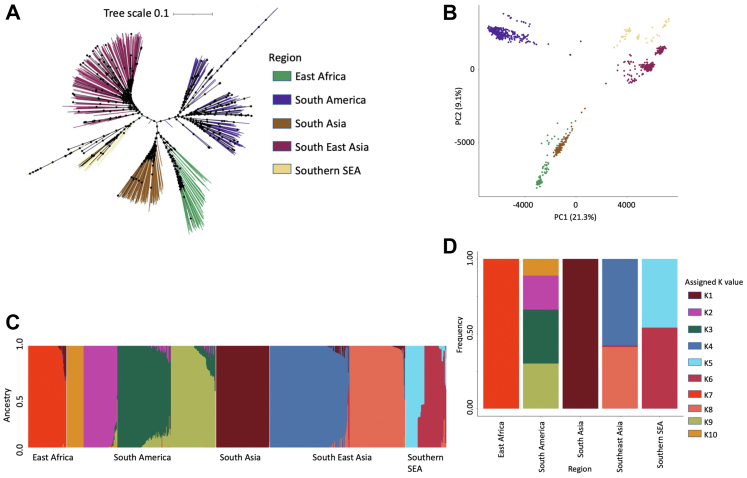
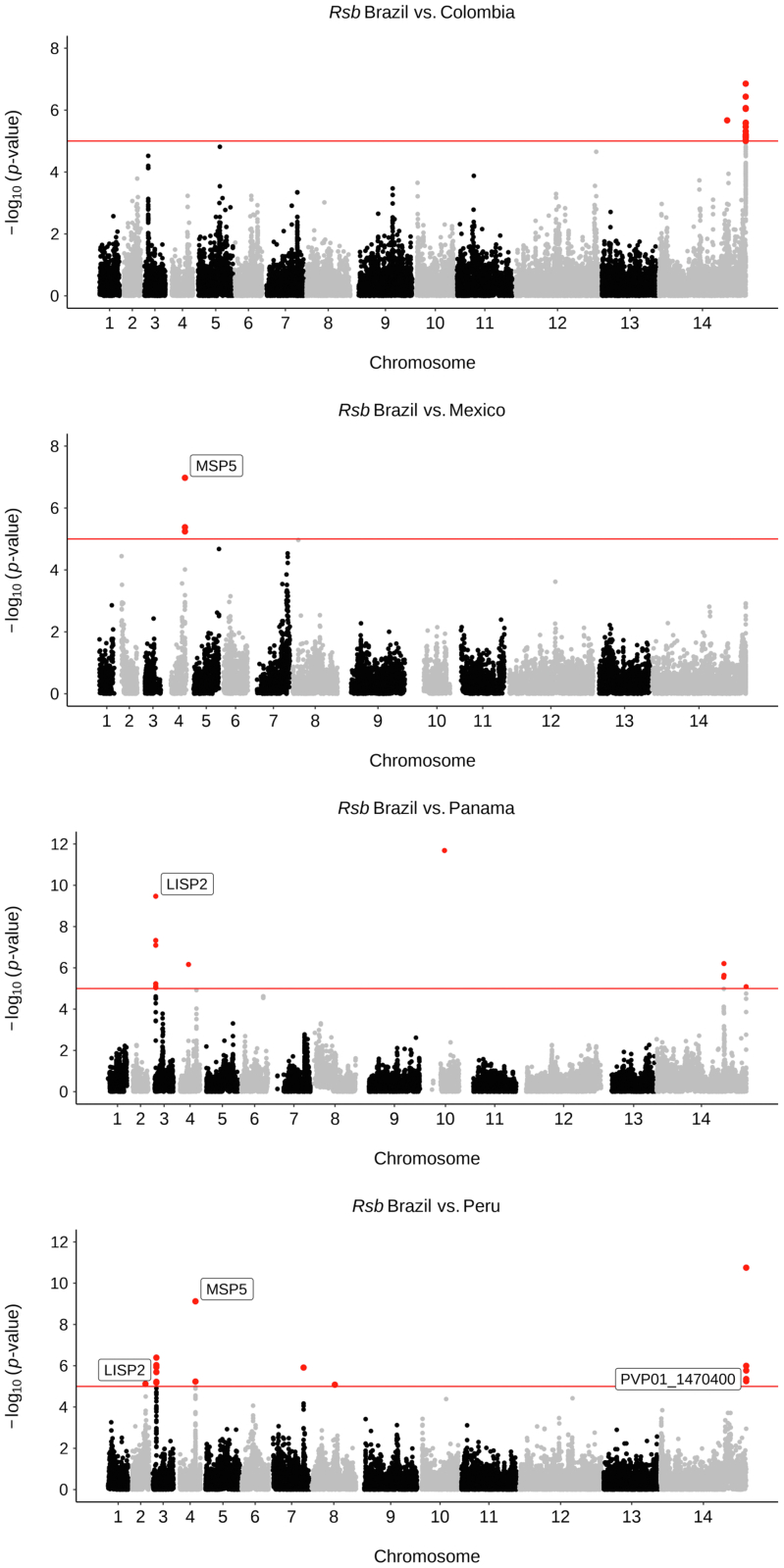
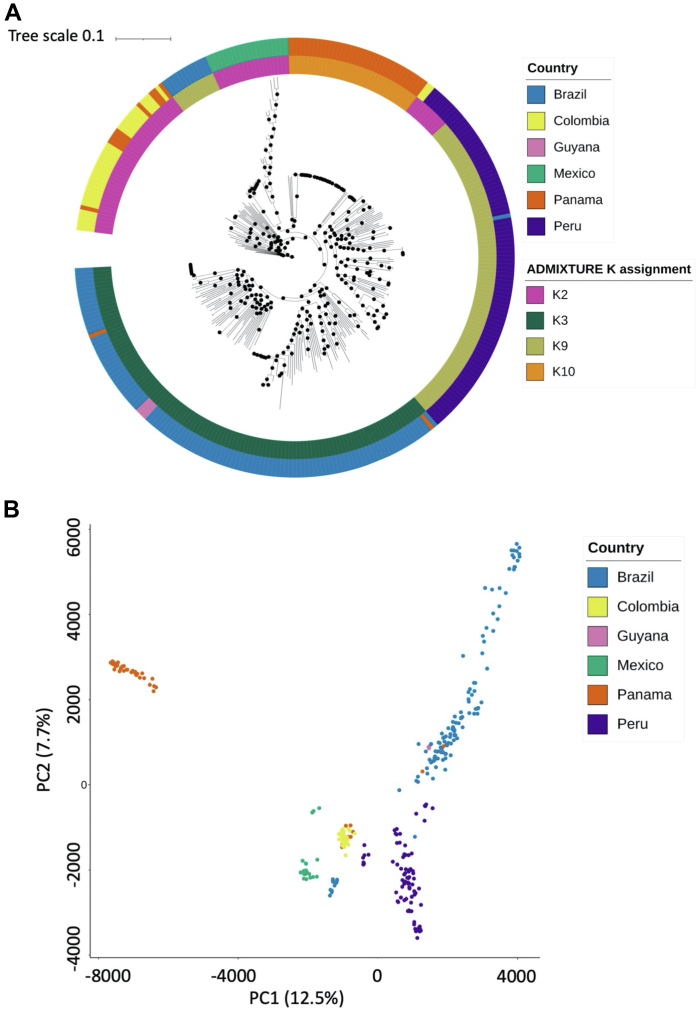
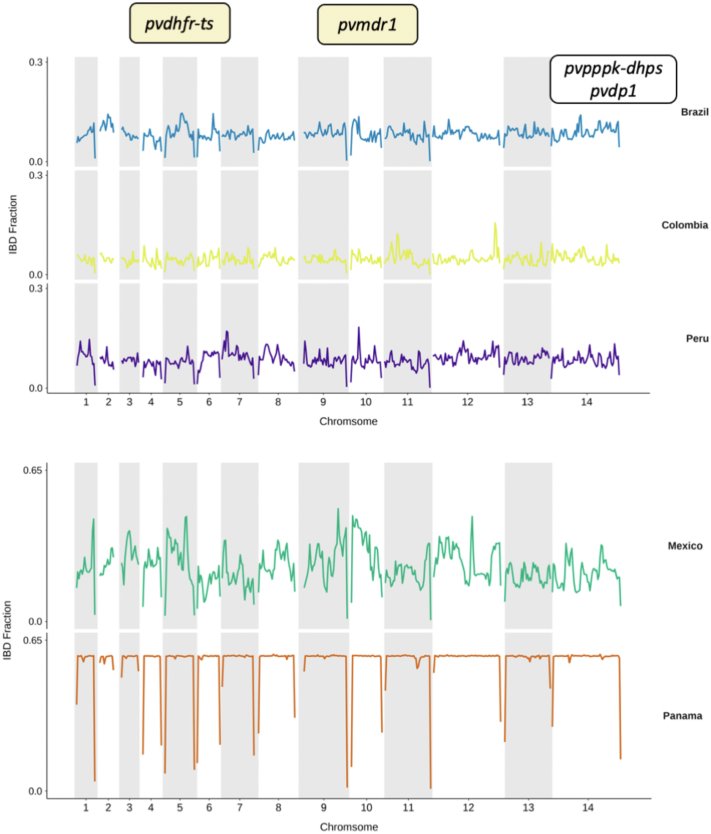
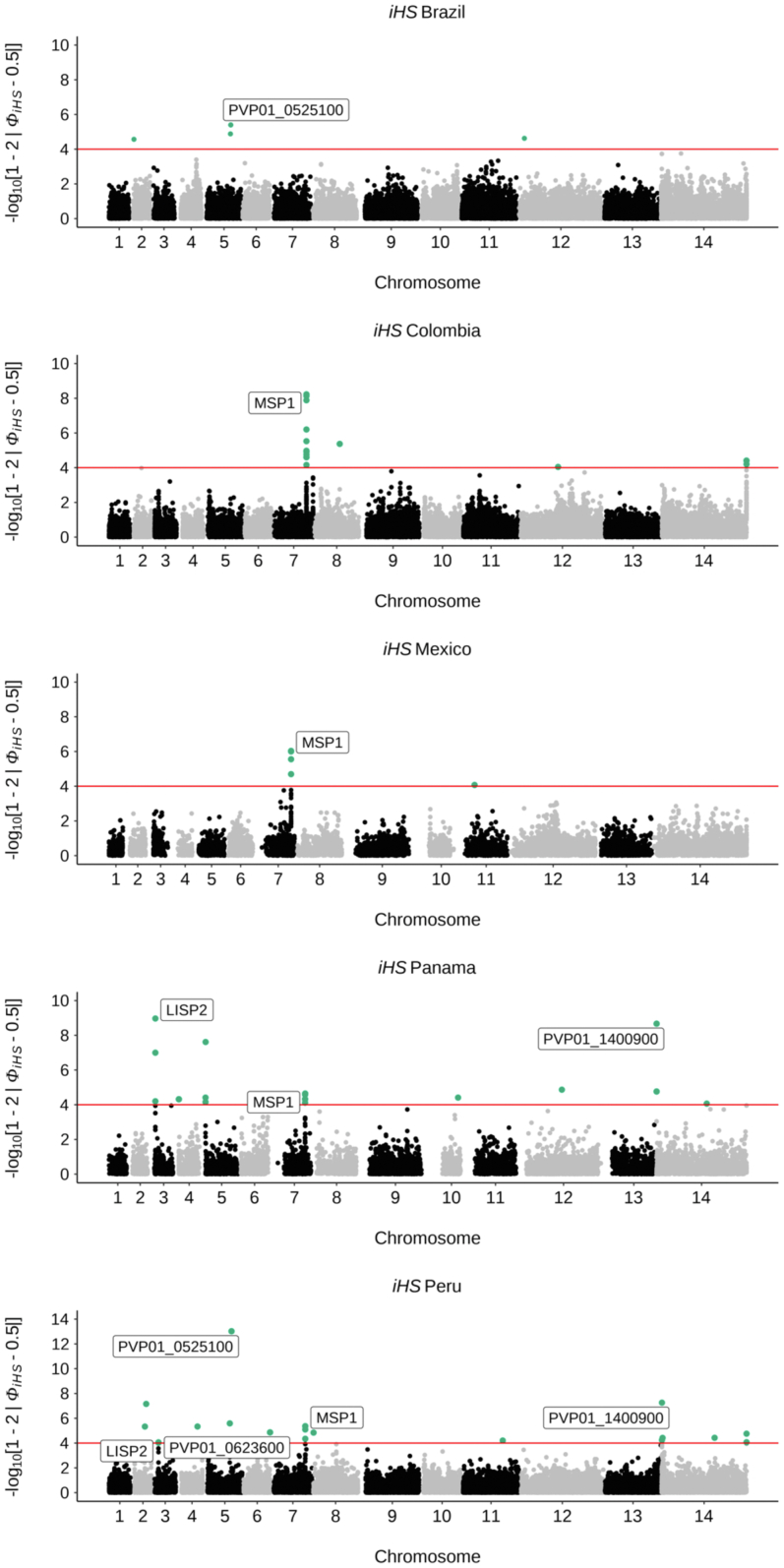
We confirm that South American isolates are distinct, have more ancestral populations than the other global regions, with differentiating mutations in genes under selective pressure linked to antimalarial drugs (, ) and mosquito vectors (). We demonstrate Brazil as a distinct parasite population, with signals of selection including ABC transporter () and PHIST exported proteins.
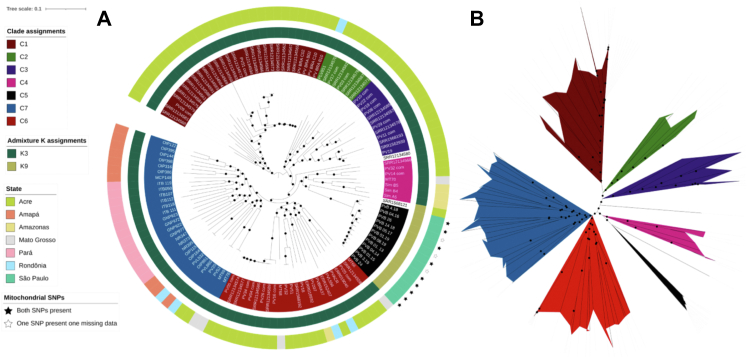
Brazil has a complex population structure, with evidence of infections and Amazonian parasites separating into multiple clusters. Overall, our work provides the first Brazil-wide analysis of population structure and identifies important mutations, which can inform future research and control measures.
AI is funded by an MRC LiD PhD studentship. TGC is funded by the Medical Research Council (Grant no. MR/M01360X/1, MR/N010469/1, MR/R025576/1, MR/R020973/1 and MR/X005895/1). SC is funded by Medical Research Council UK grants (MR/M01360X/1, MR/R025576/1, MR/R020973/1 and MR/X005895/1) and Bloomsbury SET (ref. CCF17-7779). FN is funded by The Shloklo Malaria Research Unit - part of the Mahidol Oxford Research Unit, supported by the Wellcome Trust (Grant no. 220211). ARSB is funded by São Paulo Research Foundation - FAPESP (Grant no. 2002/09546-1). RLDM is funded by Brazilian National Council for Scientific and Technological Development - CNPq (Grant no. 302353/2003-8 and 471605/2011-5); CRFM is funded by FAPESP (Grant no. 2020/06747-4) and CNPq (Grant no. 302917/2019-5 and 408636/2018-1); JGD is funded by FAPESP fellowships (2016/13465-0 and 2019/12068-5) and CNPq (Grant no. 409216/2018-6).
巴西是一个独特且研究不足的疟疾流行地区,其复杂的传播疫源地与人类和环境状况相关。了解巴西各地疟原虫的种群基因组多样性有助于支持疟疾控制策略。
通过对巴西7个州的分离株进行全基因组测序,我们采用种群基因组方法比较了巴西国内(n = 123)、各大洲(6个国家,n = 315)以及全球范围(26个国家,n = 885)内的遗传多样性。
我们证实南美分离株具有独特性,比其他全球区域拥有更多的祖先种群,在与抗疟药物(,)和蚊媒()相关的选择压力下,基因中存在分化突变。我们证明巴西是一个独特的疟原虫种群,具有选择信号,包括ABC转运蛋白()和PHIST输出蛋白。
巴西具有复杂的种群结构,有证据表明感染和亚马逊疟原虫分为多个簇。总体而言,我们的工作首次对巴西范围内的种群结构进行了分析,并确定了重要突变,可为未来的研究和控制措施提供参考。
AI由医学研究委员会(MRC)的LiD博士奖学金资助。TGC由医学研究委员会资助(资助编号:MR/M01360X/1、MR/N010469/1、MR/R025576/1、MR/R020973/1和MR/X005895/1)。SC由英国医学研究委员会资助(MR/M01360X/1、MR/R025576/1、MR/R020973/1和MR/X005895/1)以及布鲁姆斯伯里SET(参考文献CCF17 - 7779)。FN由Shloklo疟疾研究单位资助,该单位是玛希隆牛津研究单位的一部分,由惠康信托基金支持(资助编号:220211)。ARSB由圣保罗研究基金会 - FAPESP资助(资助编号:2002/09546 - 1)。RLDM由巴西国家科学技术发展委员会 - CNPq资助(资助编号:302353/2003 - 8和471605/2011 - 5);CRFM由FAPESP资助(资助编号:2020/06747 - 4)和CNPq资助(资助编号:302917/2019 - 5和408636/2018 - 1);JGD由FAPESP奖学金资助(2016/13465 - 0和2019/12068 - 5)以及CNPq资助(资助编号:409216/2018 - 6)。