Gregor Mendel Institute, Austrian Academy of Sciences, Vienna Biocenter, Vienna, Austria.
Department of Plant Sciences, University of Cambridge, Cambridge, UK.
Genome Biol. 2023 Mar 9;24(1):44. doi: 10.1186/s13059-023-02875-3.
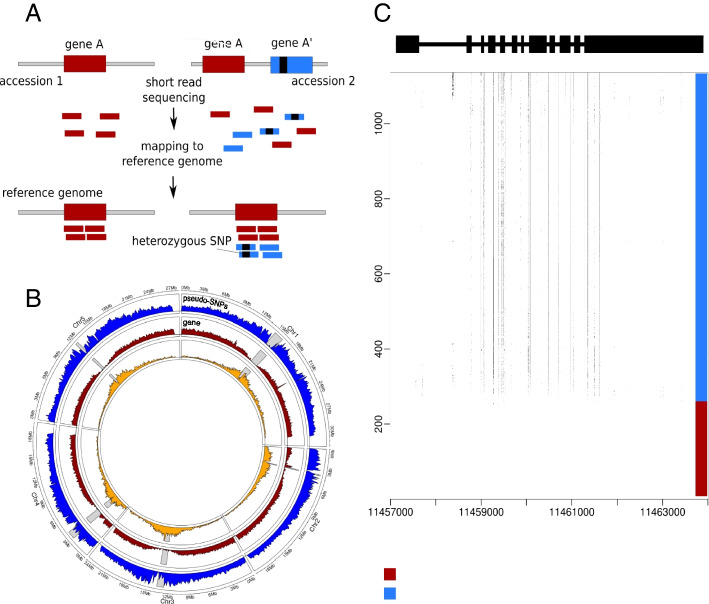
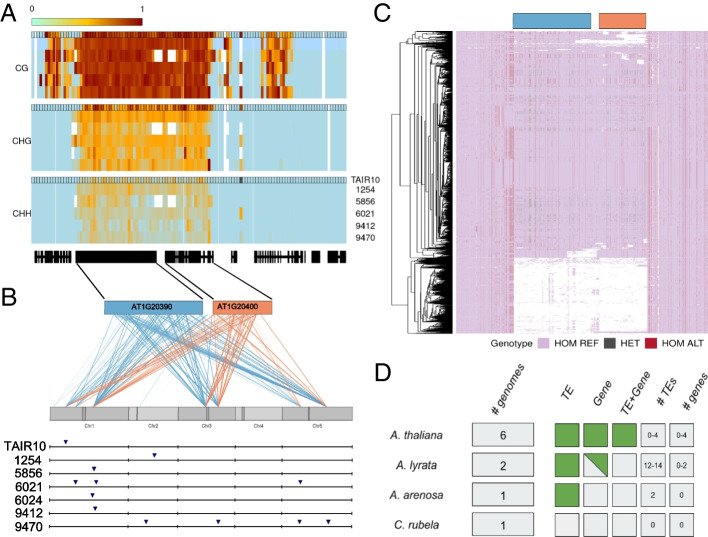
It is apparent that genomes harbor much structural variation that is largely undetected for technical reasons. Such variation can cause artifacts when short-read sequencing data are mapped to a reference genome. Spurious SNPs may result from mapping of reads to unrecognized duplicated regions. Calling SNP using the raw reads of the 1001 Arabidopsis Genomes Project we identified 3.3 million (44%) heterozygous SNPs. Given that Arabidopsis thaliana (A. thaliana) is highly selfing, and that extensively heterozygous individuals have been removed, we hypothesize that these SNPs reflected cryptic copy number variation.
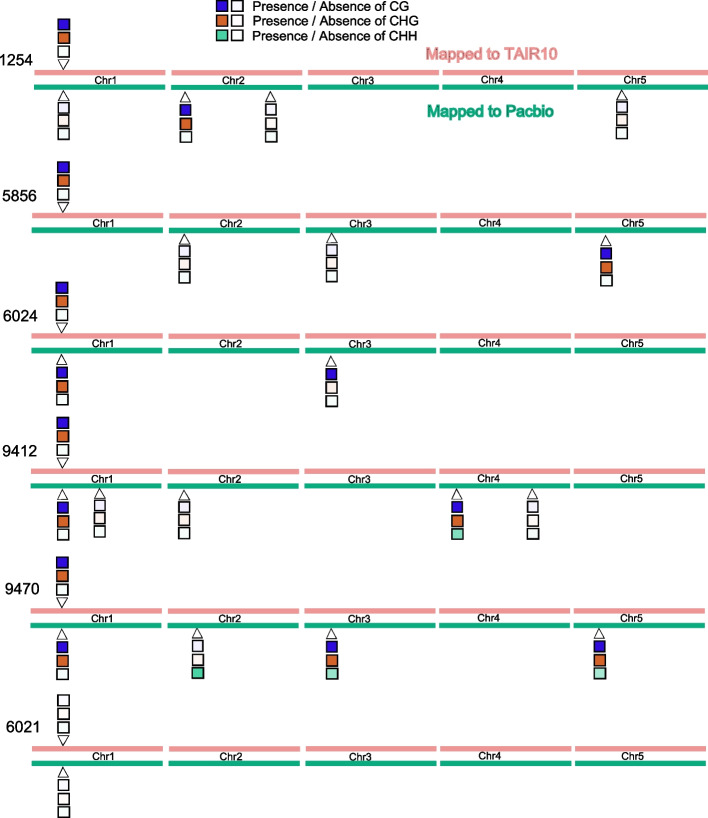
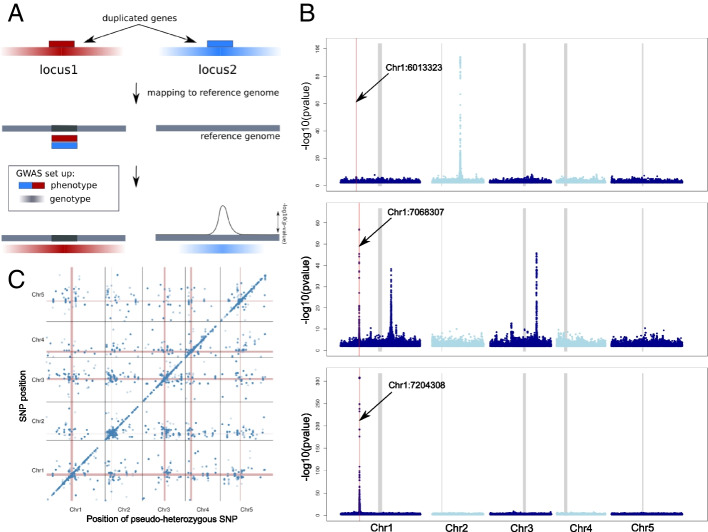
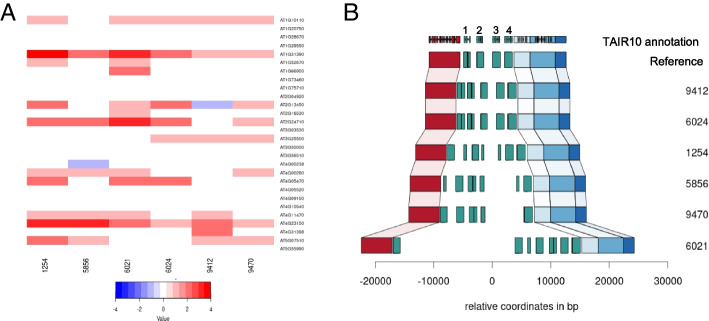
The heterozygosity we observe consists of particular SNPs being heterozygous across individuals in a manner that strongly suggests it reflects shared segregating duplications rather than random tracts of residual heterozygosity due to occasional outcrossing. Focusing on such pseudo-heterozygosity in annotated genes, we use genome-wide association to map the position of the duplicates. We identify 2500 putatively duplicated genes and validate them using de novo genome assemblies from six lines. Specific examples included an annotated gene and nearby transposon that transpose together. We also demonstrate that cryptic structural variation produces highly inaccurate estimates of DNA methylation polymorphism.
Our study confirms that most heterozygous SNP calls in A. thaliana are artifacts and suggest that great caution is needed when analyzing SNP data from short-read sequencing. The finding that 10% of annotated genes exhibit copy-number variation, and the realization that neither gene- nor transposon-annotation necessarily tells us what is actually mobile in the genome suggests that future analyses based on independently assembled genomes will be very informative.
显然,基因组中存在大量结构变异,这些变异在很大程度上由于技术原因而未被检测到。当短读测序数据映射到参考基因组时,这种变异可能会导致伪 SNP。读取映射到未识别的重复区域可能会导致伪 SNP 的产生。使用 1001 拟南芥基因组计划的原始读取进行 SNP 调用,我们鉴定出 330 万个(44%)杂合 SNP。由于拟南芥(A. thaliana)高度自交,并且已经去除了广泛的杂合个体,我们假设这些 SNP 反映了隐性拷贝数变异。
我们观察到的杂合性由个体之间特定 SNP 杂合的方式组成,这强烈表明它反映了共享的分离重复,而不是由于偶尔的杂交而导致的随机剩余杂合性的轨迹。我们专注于注释基因中的这种假杂合性,使用全基因组关联来映射重复的位置。我们鉴定出 2500 个可能的重复基因,并使用来自六个品系的从头基因组组装来验证它们。具体例子包括一个注释基因和附近一起转座的转座子。我们还证明了隐性结构变异会产生高度不准确的 DNA 甲基化多态性估计值。
我们的研究证实,拟南芥中大多数杂合 SNP 调用都是伪 SNP,并表明在分析短读测序的 SNP 数据时需要非常谨慎。发现 10%的注释基因表现出拷贝数变异,并且认识到基因和转座子注释不一定能告诉我们基因组中哪些是实际可移动的,这表明基于独立组装基因组的未来分析将非常有启发性。