Fischer Martin C, Rellstab Christian, Leuzinger Marianne, Roumet Marie, Gugerli Felix, Shimizu Kentaro K, Holderegger Rolf, Widmer Alex
ETH Zürich, Institute of Integrative Biology, Universitätstrasse 16, 8092, Zürich, Switzerland.
WSL Swiss Federal Research Institute, Zürcherstrasse 111, 8903, Birmensdorf, Switzerland.
BMC Genomics. 2017 Jan 11;18(1):69. doi: 10.1186/s12864-016-3459-7.
Microsatellite markers are widely used for estimating genetic diversity within and differentiation among populations. However, it has rarely been tested whether such estimates are useful proxies for genome-wide patterns of variation and differentiation. Here, we compared microsatellite variation with genome-wide single nucleotide polymorphisms (SNPs) to assess and quantify potential marker-specific biases and derive recommendations for future studies. Overall, we genotyped 180 Arabidopsis halleri individuals from nine populations using 20 microsatellite markers. Twelve of these markers were originally developed for Arabidopsis thaliana (cross-species markers) and eight for A. halleri (species-specific markers). We further characterized 2 million SNPs across the genome with a pooled whole-genome re-sequencing approach (Pool-Seq).
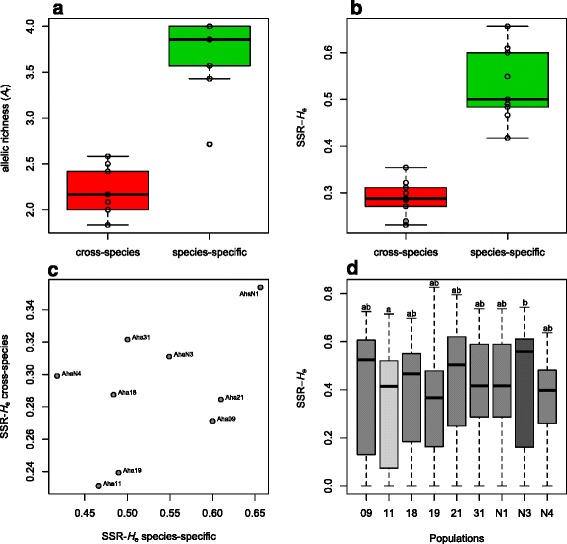
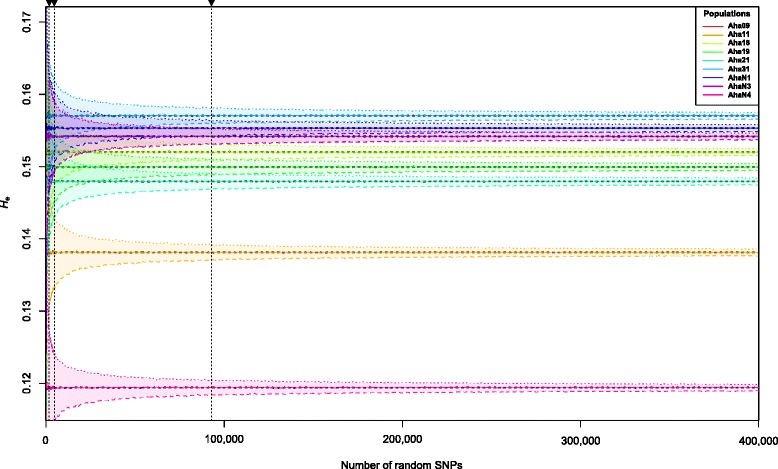
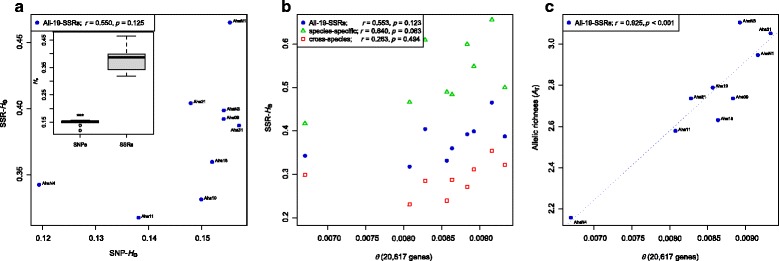
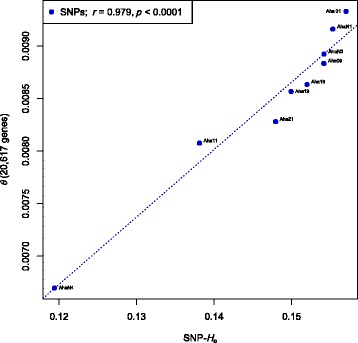
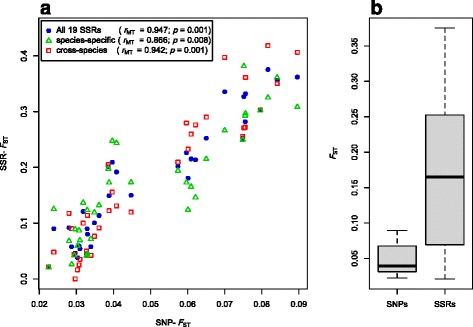
Our analyses revealed that estimates of genetic diversity and differentiation derived from cross-species and species-specific microsatellites differed substantially and that expected microsatellite heterozygosity (SSR-H ) was not significantly correlated with genome-wide SNP diversity estimates (SNP-H and θ ) in A. halleri. Instead, microsatellite allelic richness (A ) was a better proxy for genome-wide SNP diversity. Estimates of genetic differentiation among populations (F ) based on both marker types were correlated, but microsatellite-based estimates were significantly larger than those from SNPs. Possible causes include the limited number of microsatellite markers used, marker ascertainment bias, as well as the high variance in microsatellite-derived estimates. In contrast, genome-wide SNP data provided unbiased estimates of genetic diversity independent of whether genome- or only exome-wide SNPs were used. Further, we inferred that a few thousand random SNPs are sufficient to reliably estimate genome-wide diversity and to distinguish among populations differing in genetic variation.
We recommend that future analyses of genetic diversity within and differentiation among populations use randomly selected high-throughput sequencing-based SNP data to draw conclusions on genome-wide diversity patterns. In species comparable to A. halleri, a few thousand SNPs are sufficient to achieve this goal.
微卫星标记广泛用于估计种群内的遗传多样性和种群间的分化。然而,很少有人测试过这些估计是否是全基因组变异和分化模式的有效替代指标。在此,我们比较了微卫星变异与全基因组单核苷酸多态性(SNP),以评估和量化潜在的标记特异性偏差,并为未来研究提供建议。总体而言,我们使用20个微卫星标记对来自9个种群的180株拟南芥个体进行了基因分型。其中12个标记最初是为拟南芥开发的(跨物种标记),8个是为南芥开发的(物种特异性标记)。我们还使用混合全基因组重测序方法(Pool-Seq)对全基因组中的200万个SNP进行了进一步表征。
我们的分析表明,跨物种和物种特异性微卫星得出的遗传多样性和分化估计值存在显著差异,并且在南芥中,预期微卫星杂合度(SSR-H)与全基因组SNP多样性估计值(SNP-H和θ)没有显著相关性。相反,微卫星等位基因丰富度(A)是全基因组SNP多样性的更好替代指标。基于两种标记类型的种群间遗传分化估计值(F)具有相关性,但基于微卫星的估计值明显大于基于SNP的估计值。可能的原因包括所用微卫星标记数量有限、标记确定偏差以及微卫星衍生估计值的高方差。相比之下,全基因组SNP数据提供了与使用全基因组SNP还是仅外显子组SNP无关的无偏遗传多样性估计值。此外,我们推断几千个随机SNP足以可靠地估计全基因组多样性,并区分遗传变异不同的种群。
我们建议,未来对种群内遗传多样性和种群间分化的分析应使用随机选择的基于高通量测序的SNP数据,以得出全基因组多样性模式的结论。在与南芥类似的物种中,几千个SNP足以实现这一目标。