Laboratory of Neurogenetics and Molecular Medicine - IPER, Institut de Recerca Sant Joan de Déu, 08950, Barcelona, Spain.
Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII, 08950, Barcelona, Spain.
Biol Open. 2023 Apr 15;12(4). doi: 10.1242/bio.059707. Epub 2023 Apr 3.
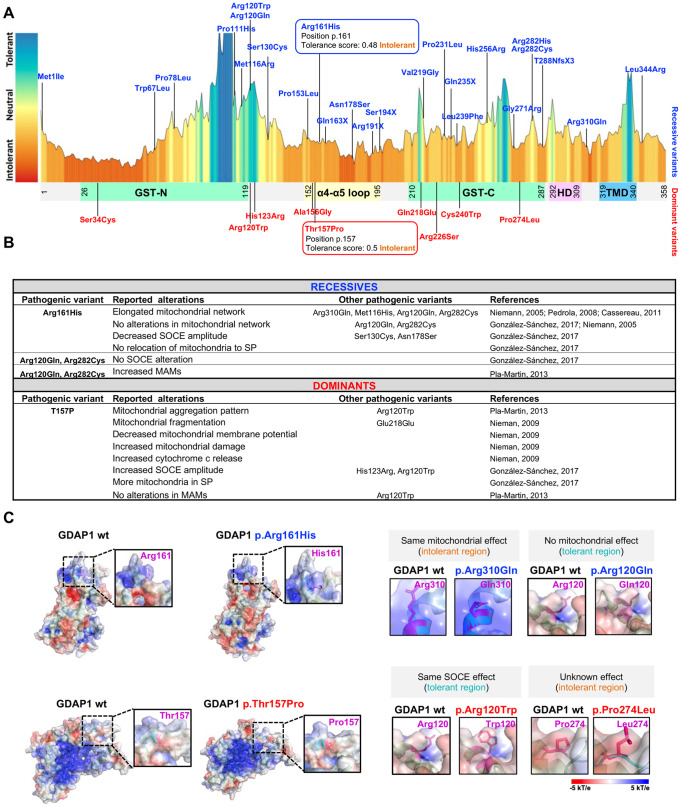
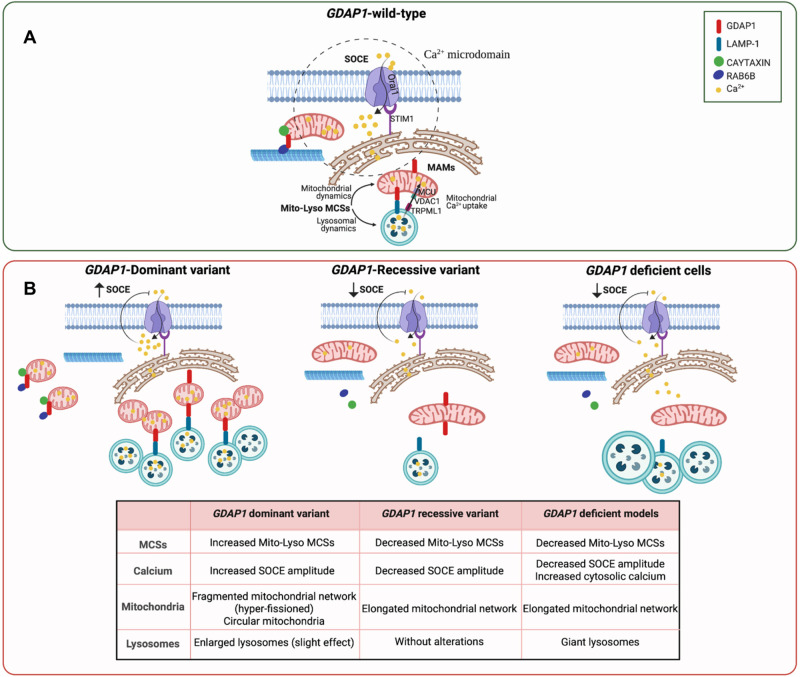
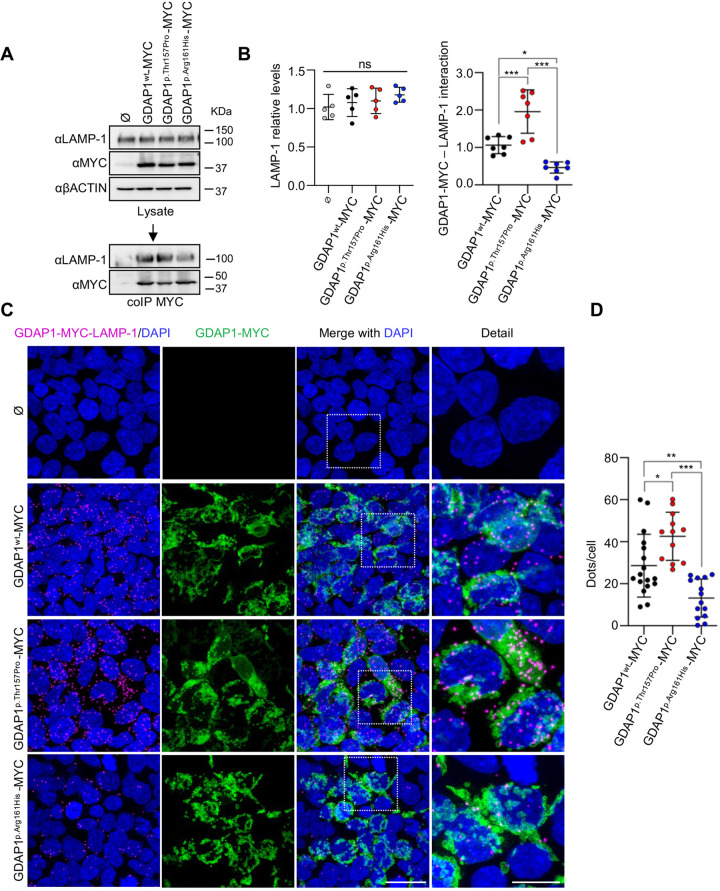
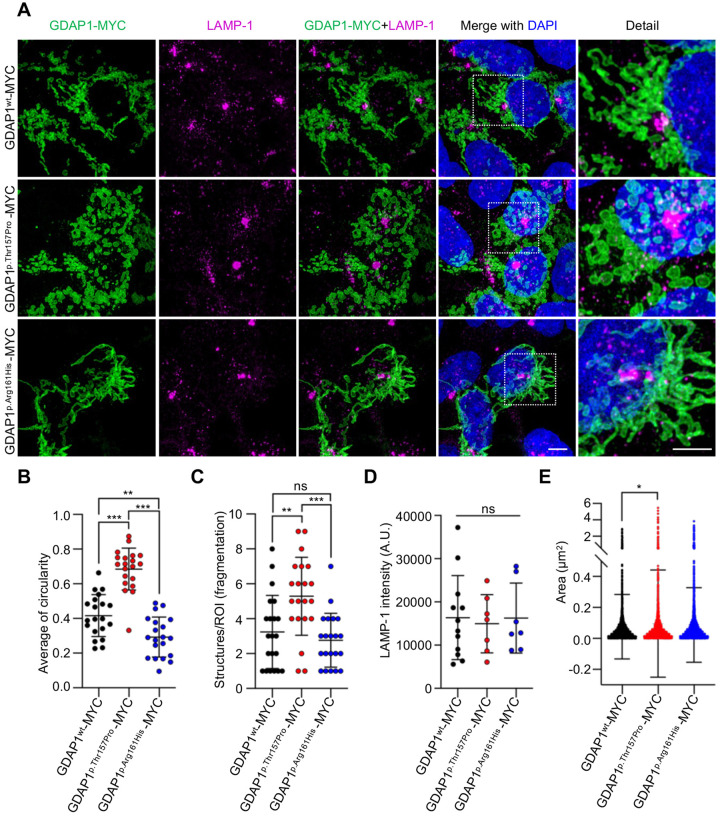
GDAP1 pathogenic variants cause Charcot-Marie-Tooth (CMT) disease, the most common hereditary motor and sensory neuropathy. CMT-GDAP1 can be axonal or demyelinating, with autosomal dominant or recessive inheritance, leading to phenotypic heterogeneity. Recessive GDAP1 variants cause a severe phenotype, whereas dominant variants are associated with a milder disease course. GDAP1 is an outer mitochondrial membrane protein involved in mitochondrial membrane contact sites (MCSs) with the plasmatic membrane, the endoplasmic reticulum (ER), and lysosomes. In GDAP1-deficient models, the pathophysiology includes morphological defects in mitochondrial network and ER, impaired Ca2+ homeostasis, oxidative stress, and mitochondrial MCSs defects. Nevertheless, the underlying pathophysiology of dominant variants is less understood. Here, we study the effect upon mitochondria-lysosome MCSs of two GDAP1 clinical variants located in the α-loop interaction domain of the protein. p.Thr157Pro dominant variant causes the increase in these MCSs that correlates with a hyper-fissioned mitochondrial network. In contrast, p.Arg161His recessive variant, which is predicted to significantly change the contact surface of GDAP1, causes decreased contacts with more elongated mitochondria. Given that mitochondria-lysosome MCSs regulate Ca2+ transfer from the lysosome to mitochondria, our results support that GDAP1 clinical variants have different consequences for Ca2+ handling and that could be primary insults determining differences in severity between dominant and recessive forms of the disease.
GDAP1 致病变体可导致夏科-马里-图什病(CMT),这是最常见的遗传性运动感觉神经病。CMT-GDAP1 可以是轴索性或脱髓鞘性的,具有常染色体显性或隐性遗传,导致表型异质性。隐性 GDAP1 变体导致严重的表型,而显性变体与更温和的疾病过程相关。GDAP1 是一种位于线粒体膜接触部位(MCSs)的外线粒体膜蛋白,与质膜、内质网(ER)和溶酶体相关。在 GDAP1 缺陷模型中,病理生理学包括线粒体网络和 ER 的形态缺陷、Ca2+ 稳态失调、氧化应激和线粒体 MCSs 缺陷。然而,对显性变体的潜在病理生理学了解较少。在这里,我们研究了位于蛋白质的α环相互作用域中的两种 GDAP1 临床变体对线粒体-溶酶体 MCSs 的影响。p.Thr157Pro 显性变体导致这些 MCSs 的增加,与线粒体超裂变网络相关。相比之下,p.Arg161His 隐性变体,预计会显著改变 GDAP1 的接触表面,导致与更长的线粒体接触减少。鉴于线粒体-溶酶体 MCSs 调节从溶酶体到线粒体的 Ca2+ 转移,我们的结果支持 GDAP1 临床变体对 Ca2+ 处理有不同的影响,并且可能是决定显性和隐性疾病形式严重程度差异的主要诱因。