Li Sanming, Roy Ethan R, Wang Yanyu, Watkins Trent, Cao Wei
Department of Anesthesiology, Critical Care and Pain Medicine, McGovern Medical School, University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Department of Neurosurgery, Baylor College of Medicine, Houston, TX, USA. Current address: Department of Neurology, University of California at San Francisco, San Francisco, CA 94158 USA.
Res Sq. 2023 Mar 7:rs.3.rs-2617457. doi: 10.21203/rs.3.rs-2617457/v1.
Alzheimer's disease (AD) is the most prevalent form of neurodegeneration. Despite the well-established link between tau aggregation and clinical progression, the major pathways driven by this protein to intrinsically damage neurons are incompletely understood.
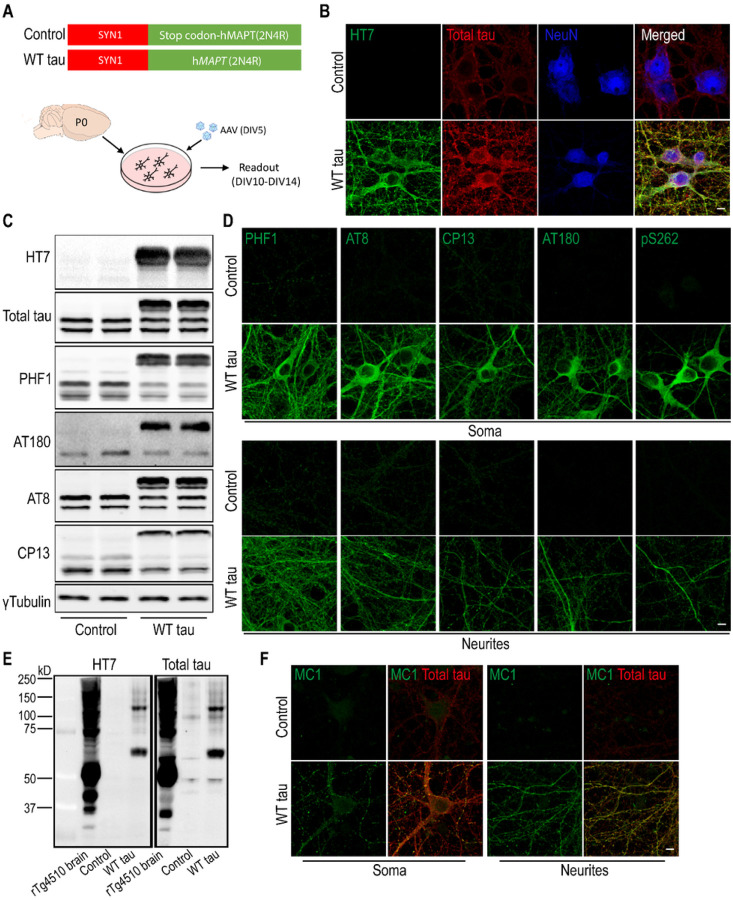
To model AD-relevant neurodegeneration driven by tau, we overexpressed wild-type human tau in primary mouse neurons and characterized the subsequent cellular and molecular changes. RNAseq profiling and functional investigation were performed as well. A direct comparison with a mutant human tau was conducted in detail.
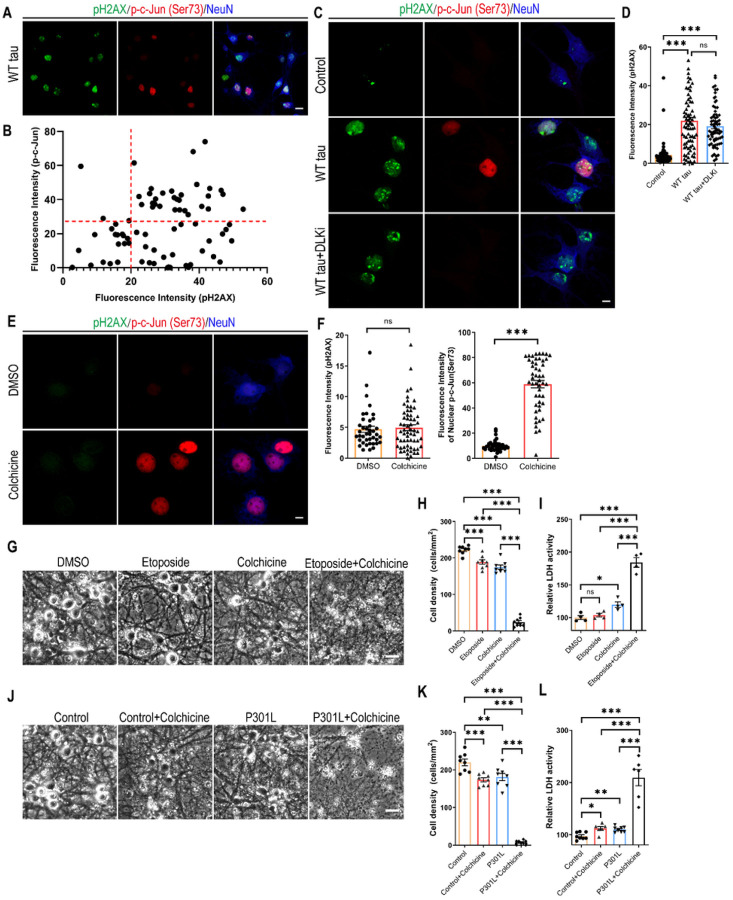
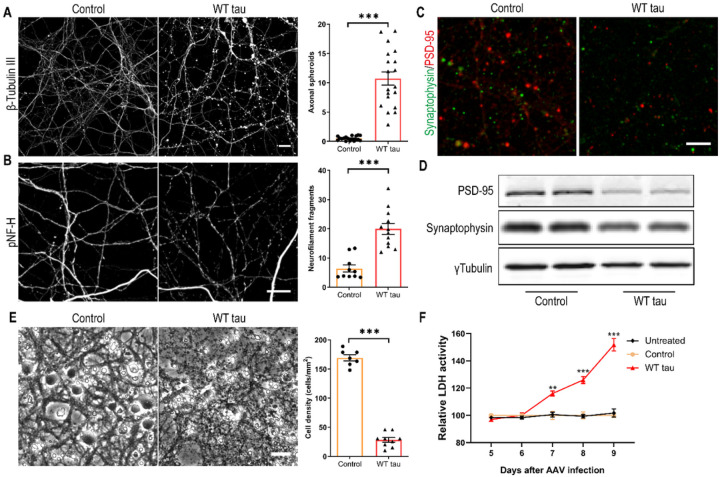
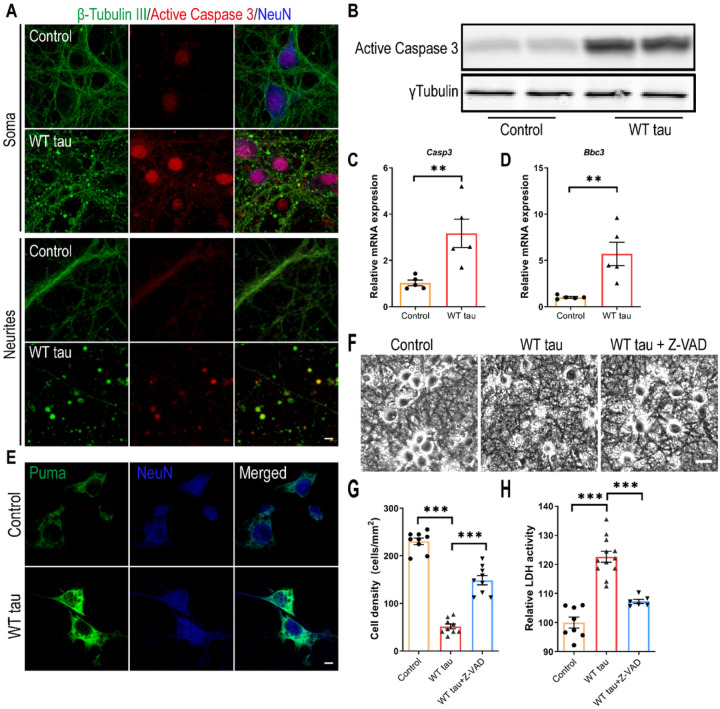
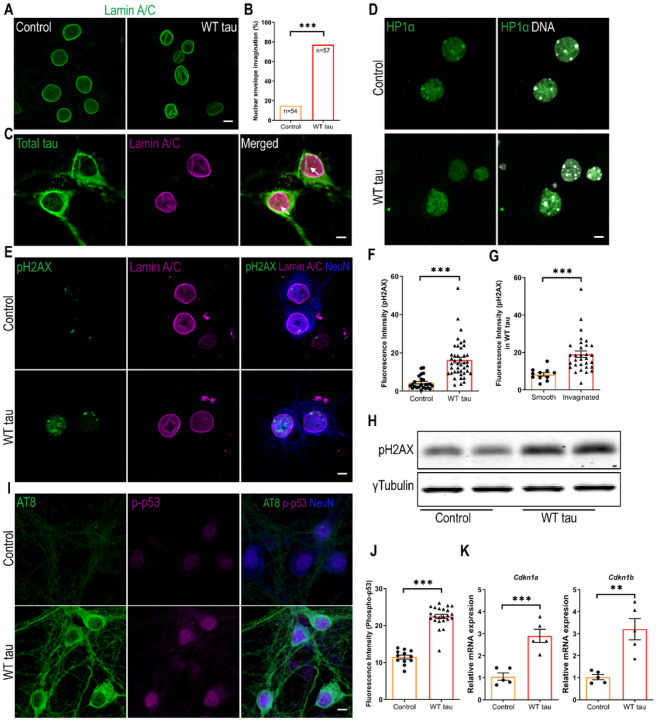
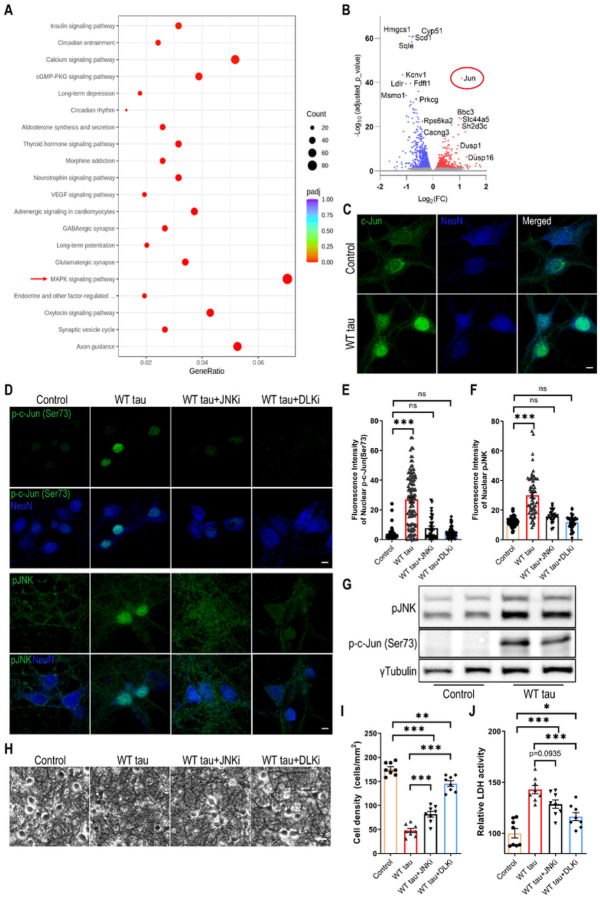
We observed substantial axonal degeneration and cell death associated with wild-type tau, a process accompanied by activated caspase 3. Mechanistically, we detected deformation of the nuclear envelope and increased DNA damage response in tau-expressing neurons. Gene profiling analysis further revealed significant alterations in the mitogen-activated protein kinase (MAPK) pathway; moreover, inhibitors of dual leucine zipper kinase (DLK) and c-Jun N-terminal kinase (JNK) were effective in alleviating wild-type human tau-induced neurodegeneration. In contrast, mutant P301L human tau was less toxic to neurons, despite causing comparable DNA damage. Axonal DLK activation induced by wild-type tau potentiated the impact of DNA damage response, resulting in overt neurotoxicity.
We have established a cellular tauopathy model highly relevant to AD and identified a functional synergy between DNA damage response and the MAPK-DLK axis in the neuronal degenerative process.
阿尔茨海默病(AD)是神经退行性变最常见的形式。尽管tau蛋白聚集与临床进展之间的联系已得到充分证实,但该蛋白导致神经元内在损伤的主要途径仍未完全明确。
为了模拟由tau蛋白驱动的与AD相关的神经退行性变,我们在原代小鼠神经元中过表达野生型人tau蛋白,并对随后的细胞和分子变化进行了表征。还进行了RNA测序分析和功能研究。并与突变型人tau蛋白进行了详细的直接比较。
我们观察到与野生型tau蛋白相关的大量轴突退变和细胞死亡,这一过程伴随着半胱天冬酶3的激活。从机制上讲,我们在表达tau蛋白的神经元中检测到核膜变形和DNA损伤反应增加。基因分析进一步揭示了丝裂原活化蛋白激酶(MAPK)途径的显著改变;此外,双亮氨酸拉链激酶(DLK)和c-Jun氨基末端激酶(JNK)的抑制剂可有效减轻野生型人tau蛋白诱导的神经退行性变。相比之下,突变型P301L人tau蛋白对神经元的毒性较小,尽管会造成相当的DNA损伤。野生型tau蛋白诱导的轴突DLK激活增强了DNA损伤反应的影响,导致明显的神经毒性。
我们建立了一个与AD高度相关的细胞tau蛋白病模型,并确定了DNA损伤反应与MAPK-DLK轴在神经元退行性变过程中的功能协同作用。