International School for Advanced Studies (SISSA), Via Bonomea 265, 34136 Trieste, Italy.
School of Information Science, Japan Advanced Institute of Science and Technology (JAIST), Asahidai 1-1, Nomi, Ishikawa 923-1292, Japan.
J Chem Theory Comput. 2023 Apr 25;19(8):2222-2229. doi: 10.1021/acs.jctc.2c01141. Epub 2023 Apr 4.
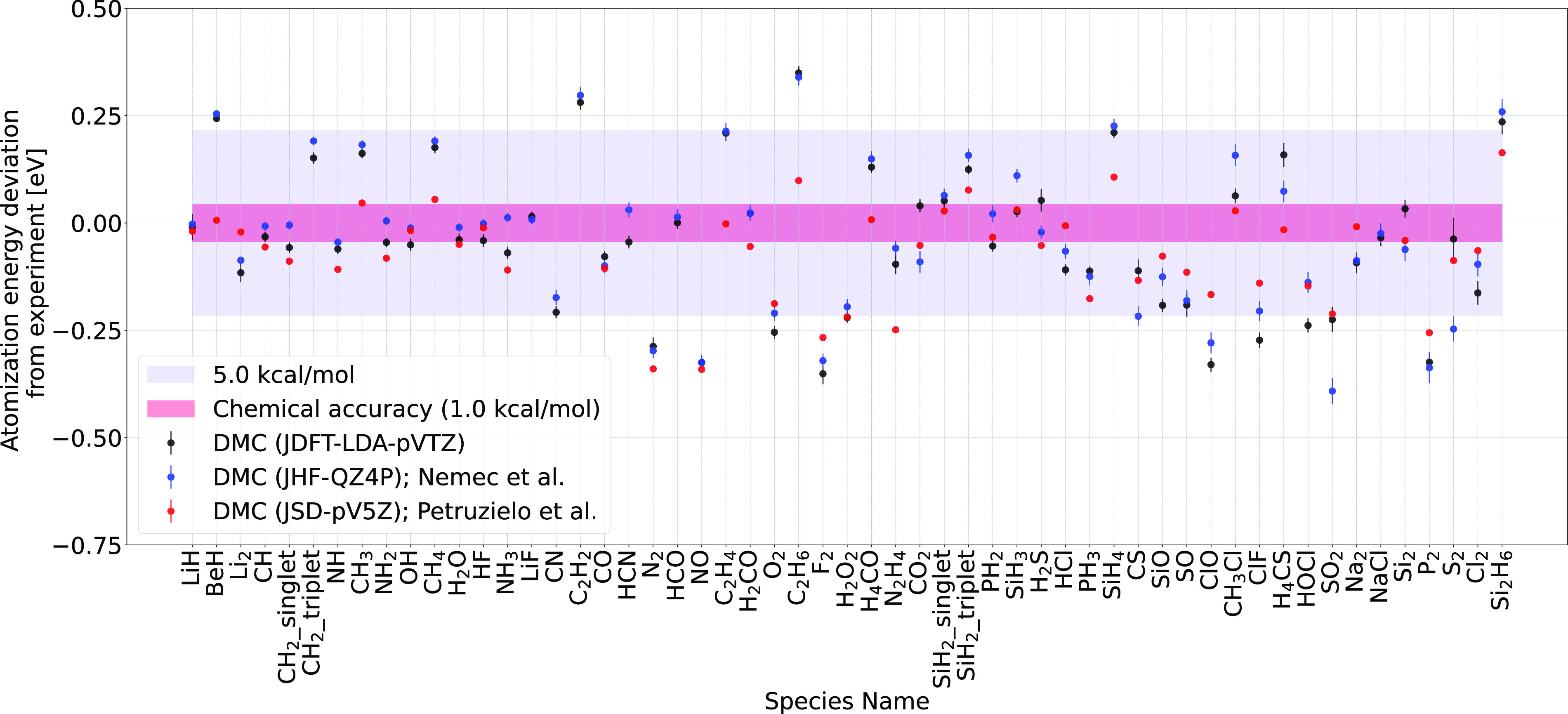
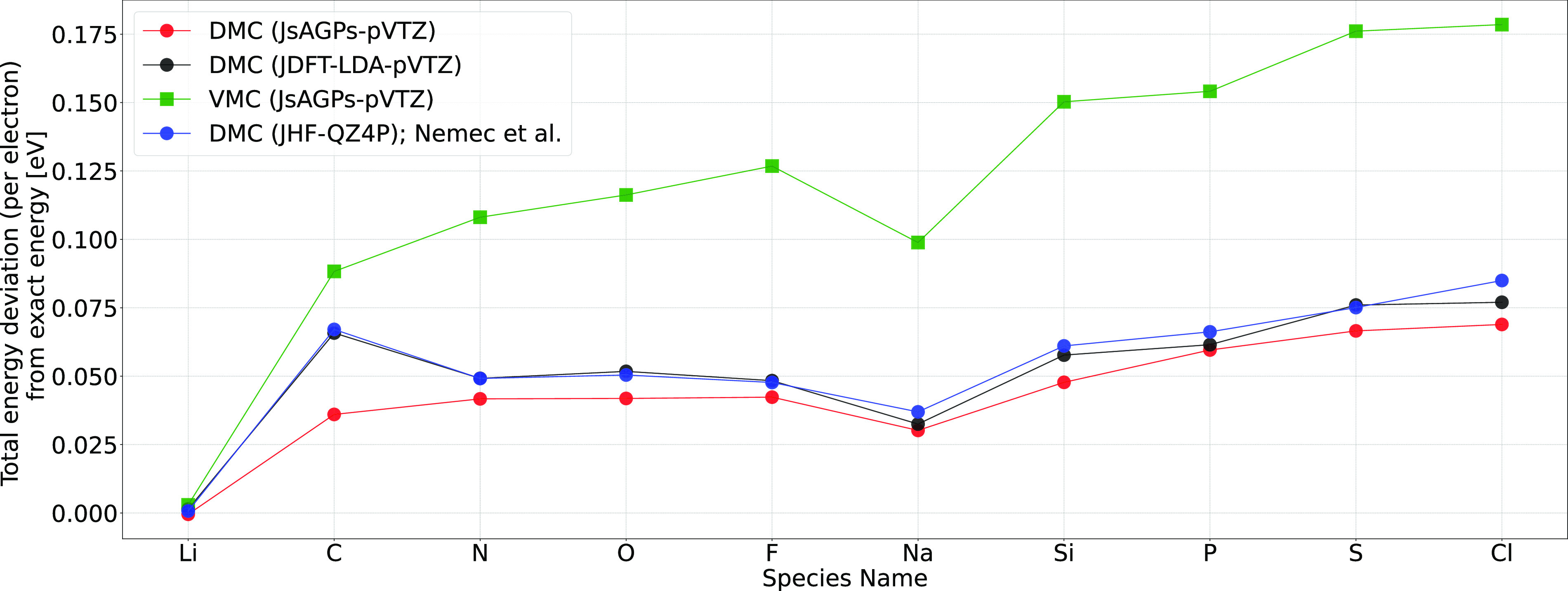
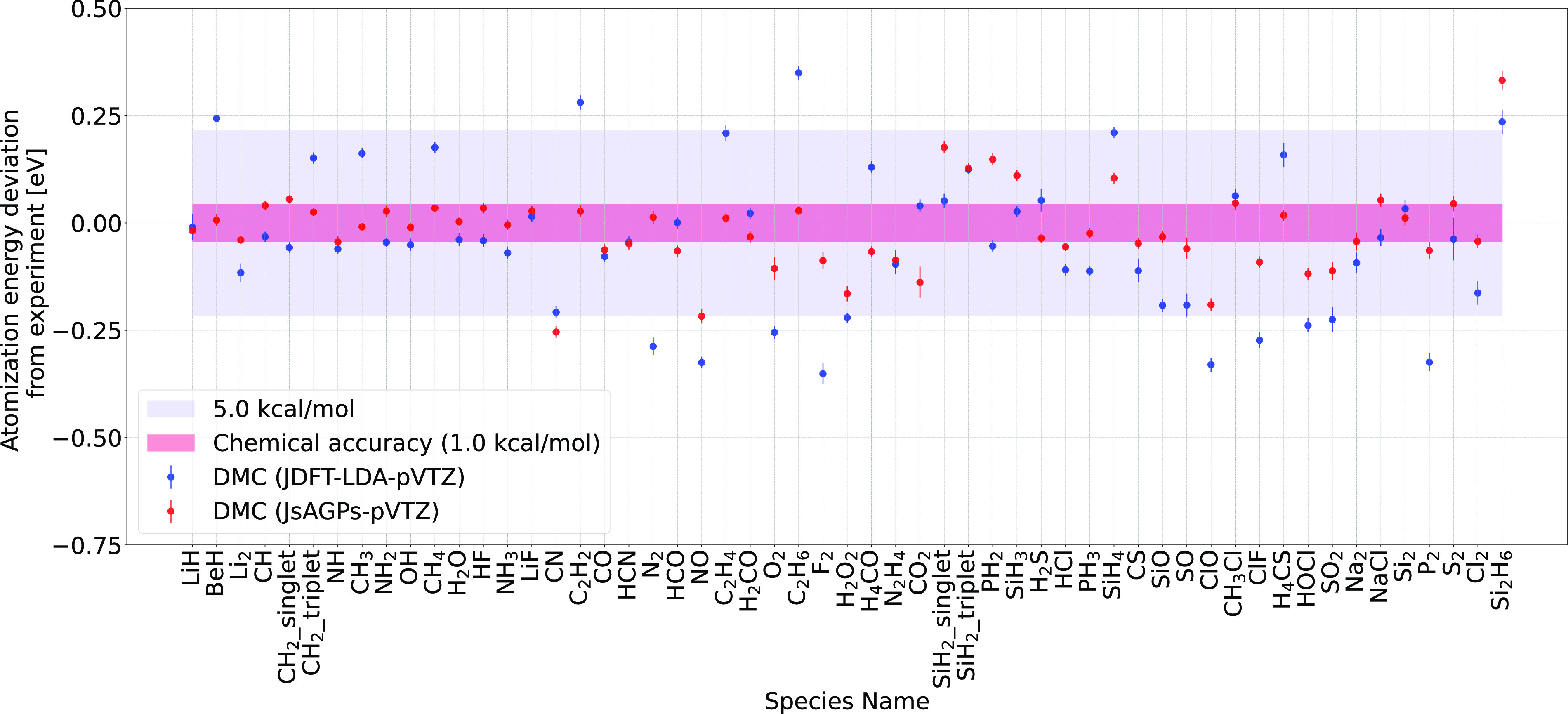
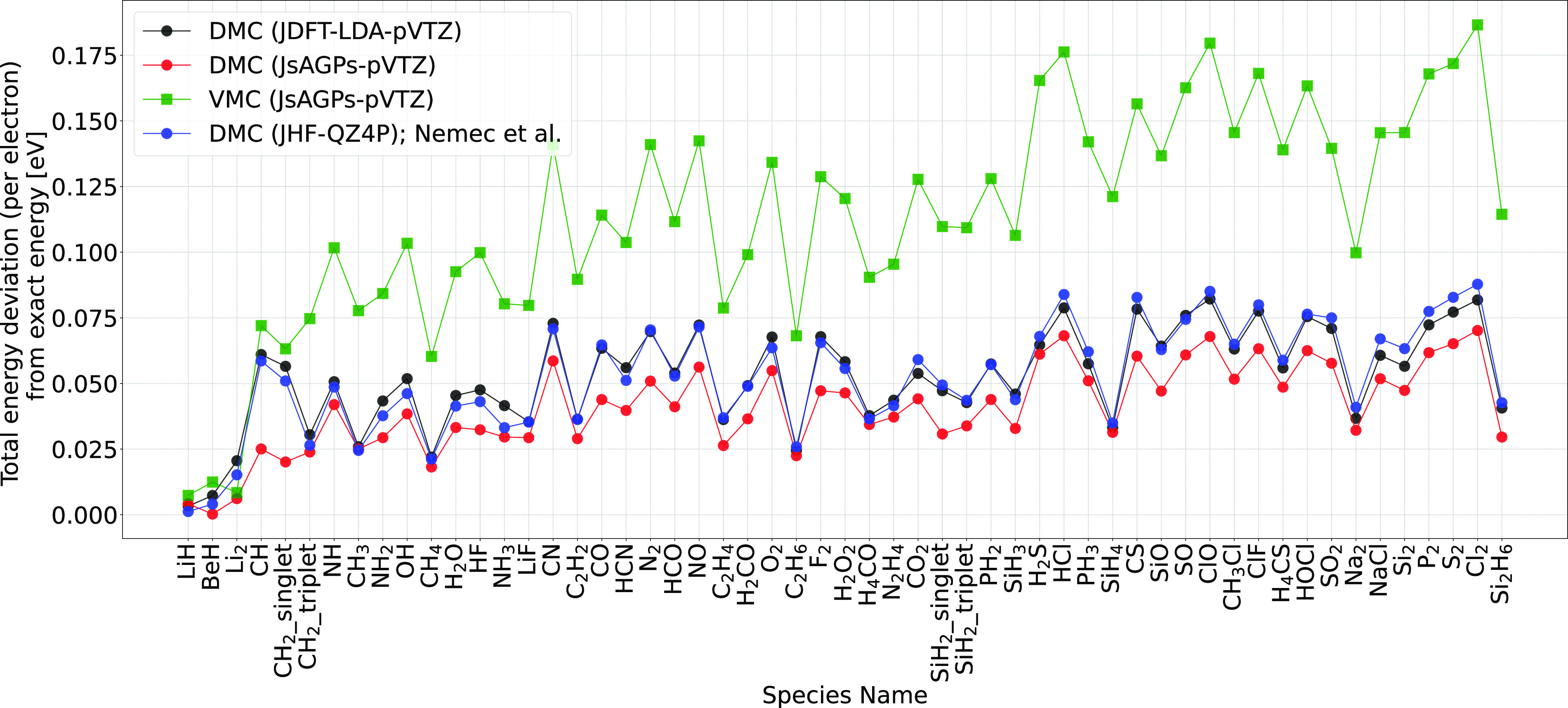
Herein, we report accurate atomization energy calculations for 55 molecules in the Gaussian-2 (G2) set using lattice regularized diffusion Monte Carlo (LRDMC). We compare the Jastrow-Slater determinant with a more flexible JsAGPs (Jastrow correlated antisymmetrized geminal power with singlet correlation) . AGPs is built from pairing functions, which explicitly include pairwise correlations among electrons, and hence, this is expected to be more efficient in recovering the correlation energy. The AGPs wave functions are first optimized at the variational Monte Carlo (VMC) level, which includes both the Jastrow factor and the nodal surface optimization. This is followed by the LRDMC projection of the . Remarkably, for many molecules, the LRDMC atomization energies obtained using the JsAGPs reach chemical accuracy (∼1 kcal/mol), and for most other molecules, the atomization energies are accurate within ∼5 kcal/mol. We obtained a mean absolute deviation of 1.6 kcal/mol with JsAGPs and 3.2 kcal/mol with JDFT (astrow factor + Slater determinant with orbitals) . This work shows the effectiveness of the flexible AGPs for atomization energy calculations and electronic structure simulations in general.
在这里,我们使用晶格正则化扩散蒙特卡罗(LRDMC)方法对高斯-2(G2)集的 55 个分子进行了精确的原子化能计算。我们将 Jastrow-Slater 行列式与更灵活的 JsAGPs(具有单重相关的 Jastrow 相关反对称双子乘积)进行了比较。AGPs 由配对函数构建,这些函数明确地包含电子之间的成对相关,因此,预计这将更有效地恢复相关能量。AGPs 波函数首先在变分蒙特卡罗(VMC)水平上进行优化,其中包括 Jastrow 因子和节面优化。然后进行 的 LRDMC 投影。值得注意的是,对于许多分子,使用 JsAGPs 得到的 LRDMC 原子化能达到化学精度(约 1 kcal/mol),对于大多数其他分子,原子化能的精度在约 5 kcal/mol 以内。我们使用 JsAGPs 获得的平均绝对偏差为 1.6 kcal/mol,使用 JDFT(包含 Jastrow 因子和 Slater 行列式的 轨道)为 3.2 kcal/mol。这项工作表明,灵活的 AGPs 对于原子化能计算和一般电子结构模拟是有效的。