Yang Yaoling, Lawson Daniel John
Department of Statistical Science, School of Mathematics, University of Bristol, Bristol BS8 1UG, UK.
Integrative Epidemiology Unit, Population Health Sciences, Bristol Medical School, University of Bristol, Bristol BS8 2BN, UK.
Bioinform Adv. 2023 Mar 23;3(1):vbad038. doi: 10.1093/bioadv/vbad038. eCollection 2023.
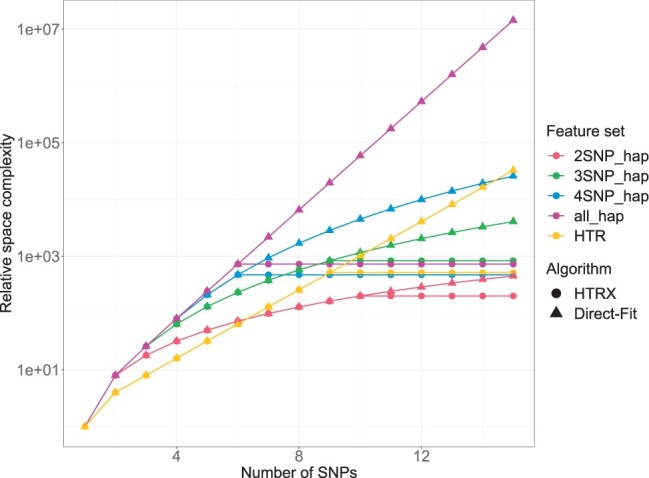
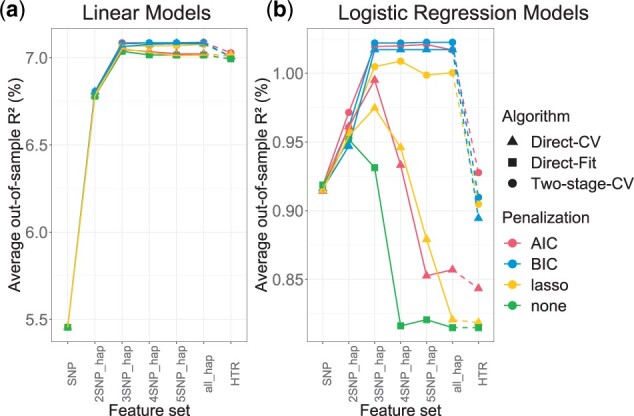
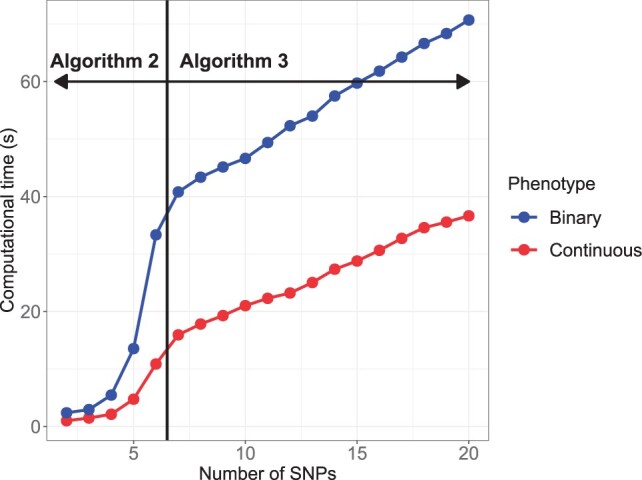
Haplotype Trend Regression with eXtra flexibility (HTRX) is an R package to learn sets of interacting features that explain variance in a phenotype. Genome-wide association studies (GWAS) have identified thousands of single nucleotide polymorphisms (SNPs) associated with complex traits and diseases, but finding the true causal signal from a high linkage disequilibrium block is challenging. We focus on the simpler task of quantifying the total variance explainable not just with main effects but also interactions and tagging, using haplotype-based associations. HTRX identifies haplotypes composed of non-contiguous SNPs associated with a phenotype and can naturally be performed on regions with a GWAS hit before or after fine-mapping. To reduce the space and computational complexity when investigating many features, we constrain the search by growing good feature sets using 'Cumulative HTRX', and limit the maximum complexity of a feature set. As the computational time scales linearly with the number of SNPs, HTRX has the potential to be applied to large chromosome regions.
HTRX is implemented in R and is available under GPL-3 licence from CRAN (https://cran.r-project.org/web/packages/HTRX/readme/README.html). The development version is maintained on GitHub (https://github.com/YaolingYang/HTRX).
Supplementary data are available at online.
具有额外灵活性的单倍型趋势回归(HTRX)是一个R包,用于学习能够解释表型变异的相互作用特征集。全基因组关联研究(GWAS)已经鉴定出数千个与复杂性状和疾病相关的单核苷酸多态性(SNP),但从高度连锁不平衡区域中找到真正的因果信号具有挑战性。我们专注于一项更简单的任务,即使用基于单倍型的关联来量化不仅可由主效应而且可由相互作用和标签解释的总变异。HTRX识别由与表型相关的非连续SNP组成的单倍型,并且可以自然地在精细定位之前或之后对具有GWAS命中的区域进行。为了在研究许多特征时降低空间和计算复杂性,我们通过使用“累积HTRX”增长良好的特征集来限制搜索,并限制特征集的最大复杂性。由于计算时间与SNP数量呈线性比例,HTRX有潜力应用于大的染色体区域。
HTRX用R实现,可在GPL-3许可下从CRAN获取(https://cran.r-project.org/web/packages/HTRX/readme/README.html)。开发版本在GitHub上维护(https://github.com/YaolingYang/HTRX)。
补充数据可在网上获取。