Division of Genetics and Genomics, Boston Children's Hospital, Harvard Medical School, Boston, MA, USA.
Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Hum Genomics. 2024 Apr 29;18(1):44. doi: 10.1186/s40246-024-00604-w.
A major obstacle faced by families with rare diseases is obtaining a genetic diagnosis. The average "diagnostic odyssey" lasts over five years and causal variants are identified in under 50%, even when capturing variants genome-wide. To aid in the interpretation and prioritization of the vast number of variants detected, computational methods are proliferating. Knowing which tools are most effective remains unclear. To evaluate the performance of computational methods, and to encourage innovation in method development, we designed a Critical Assessment of Genome Interpretation (CAGI) community challenge to place variant prioritization models head-to-head in a real-life clinical diagnostic setting.
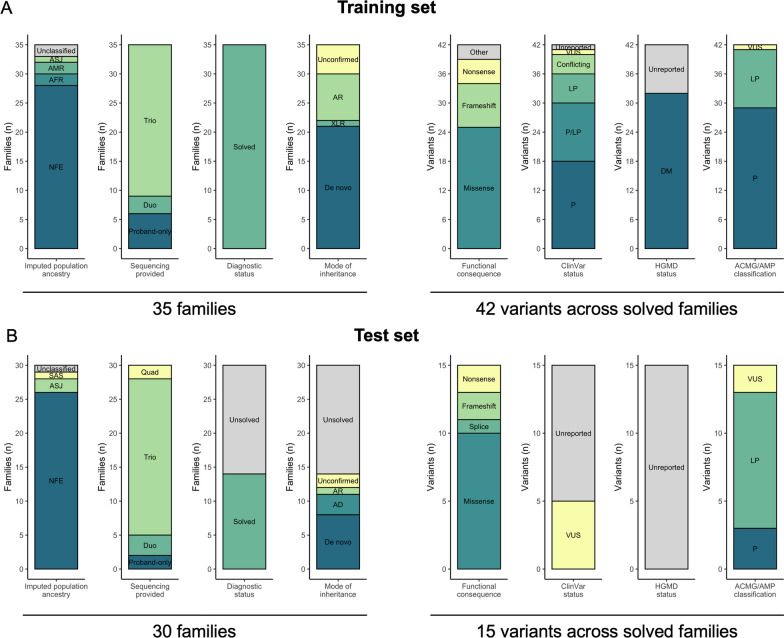
We utilized genome sequencing (GS) data from families sequenced in the Rare Genomes Project (RGP), a direct-to-participant research study on the utility of GS for rare disease diagnosis and gene discovery. Challenge predictors were provided with a dataset of variant calls and phenotype terms from 175 RGP individuals (65 families), including 35 solved training set families with causal variants specified, and 30 unlabeled test set families (14 solved, 16 unsolved). We tasked teams to identify causal variants in as many families as possible. Predictors submitted variant predictions with estimated probability of causal relationship (EPCR) values. Model performance was determined by two metrics, a weighted score based on the rank position of causal variants, and the maximum F-measure, based on precision and recall of causal variants across all EPCR values.
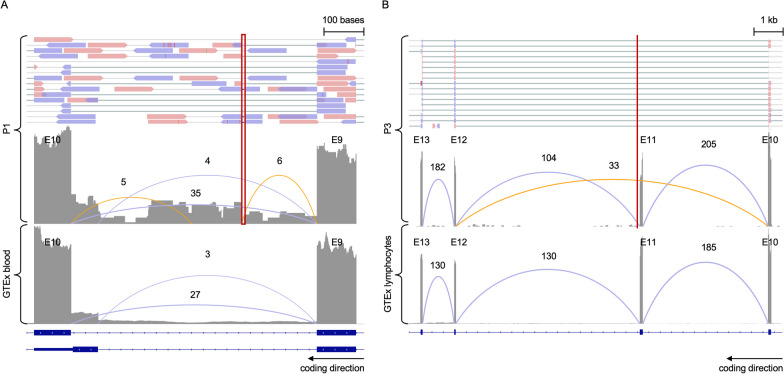
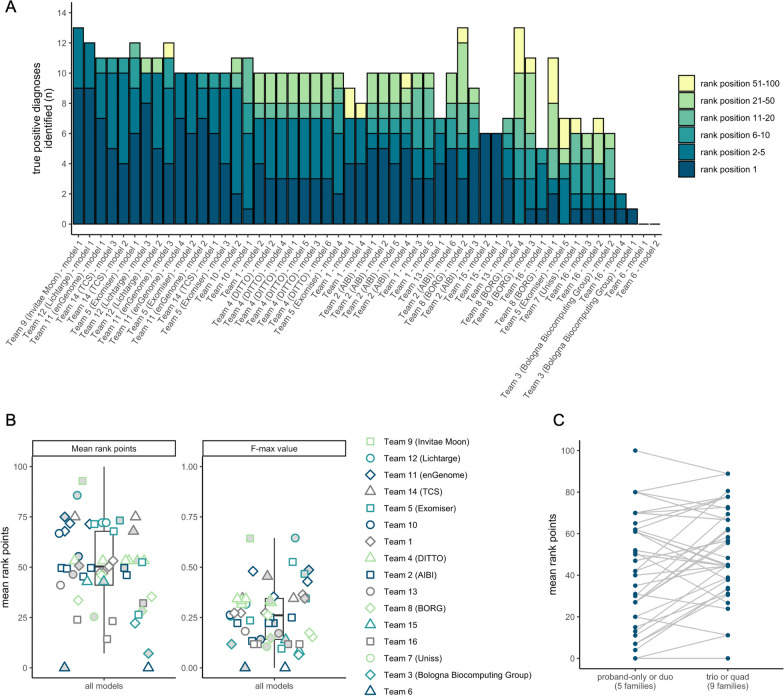
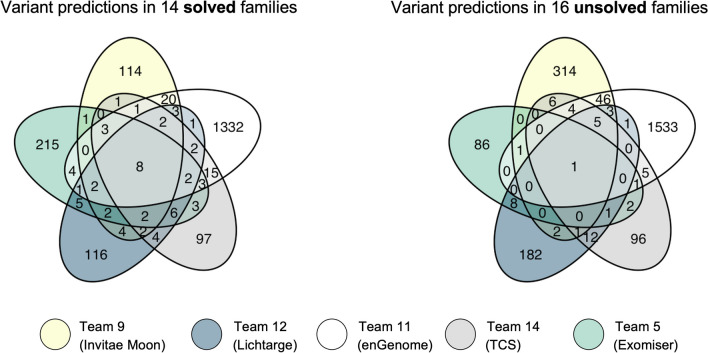
Sixteen teams submitted predictions from 52 models, some with manual review incorporated. Top performers recalled causal variants in up to 13 of 14 solved families within the top 5 ranked variants. Newly discovered diagnostic variants were returned to two previously unsolved families following confirmatory RNA sequencing, and two novel disease gene candidates were entered into Matchmaker Exchange. In one example, RNA sequencing demonstrated aberrant splicing due to a deep intronic indel in ASNS, identified in trans with a frameshift variant in an unsolved proband with phenotypes consistent with asparagine synthetase deficiency.
Model methodology and performance was highly variable. Models weighing call quality, allele frequency, predicted deleteriousness, segregation, and phenotype were effective in identifying causal variants, and models open to phenotype expansion and non-coding variants were able to capture more difficult diagnoses and discover new diagnoses. Overall, computational models can significantly aid variant prioritization. For use in diagnostics, detailed review and conservative assessment of prioritized variants against established criteria is needed.
罕见病患者家庭面临的主要障碍是获得基因诊断。平均“诊断探索”超过五年,即使在全基因组范围内捕获变异,也只有不到 50%的患者能确定病因。为了帮助解释和优先考虑大量检测到的变异,计算方法正在大量涌现。目前尚不清楚哪些工具最有效。为了评估计算方法的性能,并鼓励方法开发方面的创新,我们设计了一个基因组解读关键评估(CAGI)社区挑战,将变异优先级模型置于真实临床诊断环境中进行直接比较。
我们利用在罕见基因组项目(RGP)中对参与者进行直接测序(GS)的数据,这是一项关于 GS 在罕见病诊断和基因发现中的效用的研究。预测器提供了来自 175 名 RGP 个体(65 个家庭)的变异调用和表型术语数据集,包括 35 个具有指定因果变异的解决训练集家庭,以及 30 个未标记的测试集家庭(14 个已解决,16 个未解决)。我们的任务是尽可能多地识别出家庭中的因果变异。预测器提交了具有因果关系估计概率(EPCR)值的变异预测。模型性能通过两个指标来确定,一个是基于因果变异排名位置的加权分数,另一个是基于所有 EPCR 值的因果变异精度和召回率的最大 F 分数。
有 16 个团队提交了 52 个模型的预测结果,其中一些模型包含手动审查。表现最好的模型在排名前 5 位的变异中召回了多达 13 个因果变异。在进行了确认性 RNA 测序后,新发现的诊断性变异被返回到两个之前未解决的家庭中,两个新的疾病基因候选者被输入到 Matchmaker Exchange 中。在一个例子中,由于 ASNS 中一个深内含子插入/缺失导致的异常剪接,通过与一个未解决的先证者的移码变异在 trans 中被鉴定出来,该先证者的表型与天冬酰胺合成酶缺乏一致。
模型方法和性能差异很大。在识别因果变异时,综合考虑了变异质量、等位基因频率、预测的有害性、分离和表型的模型是有效的,而对表型扩展和非编码变异开放的模型则能够捕捉到更困难的诊断并发现新的诊断。总的来说,计算模型可以显著帮助变异优先级排序。为了在诊断中使用,需要对优先考虑的变异进行详细的审查,并根据既定标准进行保守评估。