Zeng Lu, Fujita Masashi, Gao Zongmei, White Charles C, Green Gilad S, Habib Naomi, Menon Vilas, Bennett David A, Boyle Patricia A, Klein Hans-Ulrich, De Jager Philip L
Center for Translational and Computational Neuroimmunology, Department of Neurology, Columbia University Irving Medical Center, New York, NY, USA.
Edmond & Lily Safra Center for Brain Sciences, The Hebrew University of Jerusalem, Jerusalem, Israel.
medRxiv. 2023 Mar 29:2023.03.27.23286844. doi: 10.1101/2023.03.27.23286844.
Depression is a common psychiatric illness and global public health problem. However, our limited understanding of the biological basis of depression has hindered the development of novel treatments and interventions.
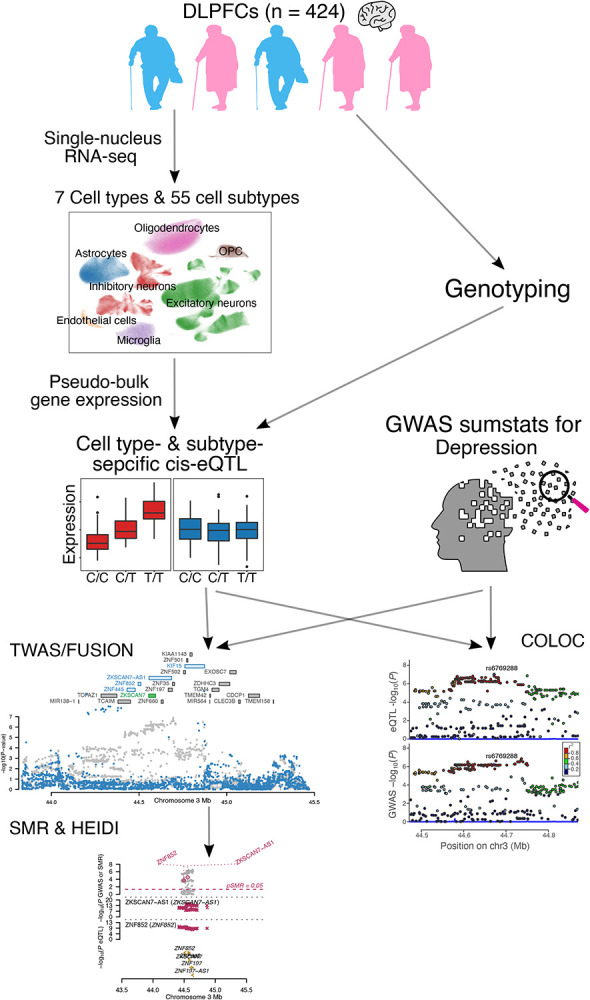
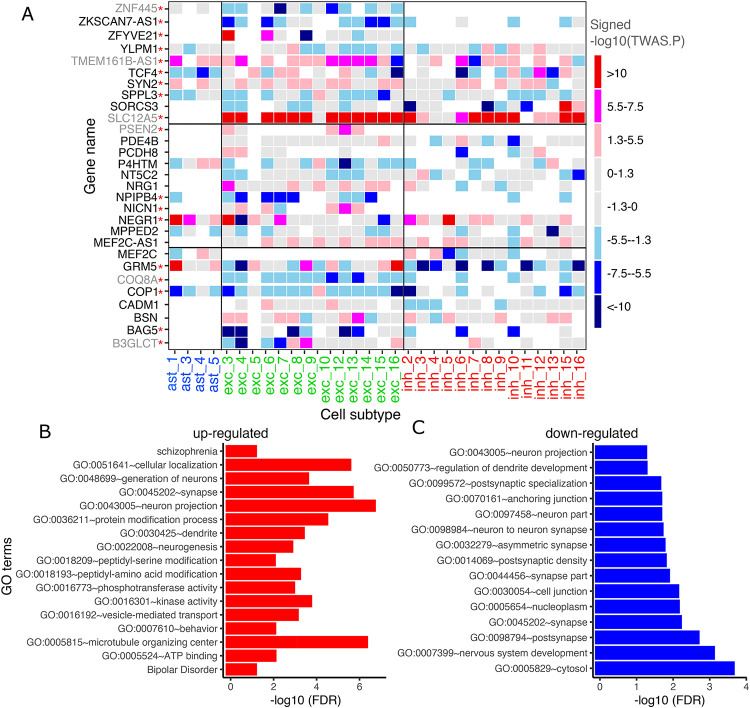
To identify new candidate genes for therapeutic development, we examined single-nucleus RNA sequencing (snucRNAseq) data from the dorsolateral prefrontal cortex (N=424) in relation to ante-mortem depressive symptoms. To complement these direct analyses, we also used genome-wide association study (GWAS) results for depression (N=500,199) along with genetic tools for inferring the expression of 22,159 genes in 7 cell types and 55 cell subtypes to perform transcriptome-wide association studies (TWAS) of depression followed by Mendelian randomization (MR).
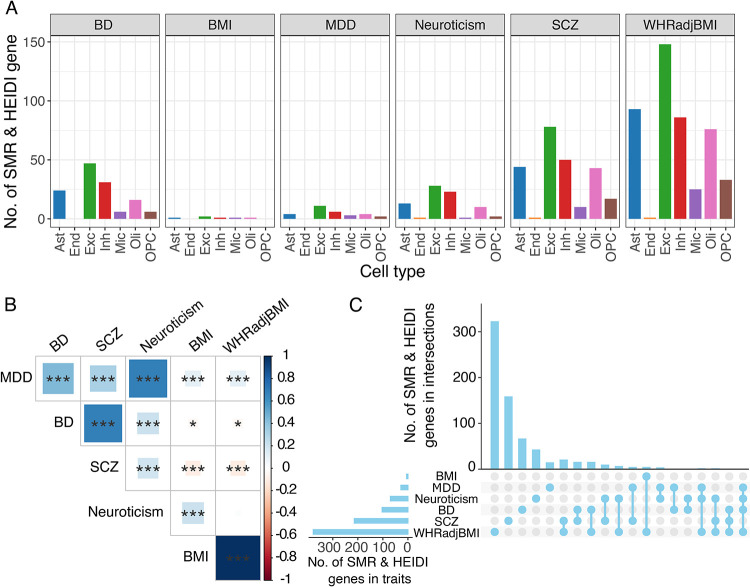
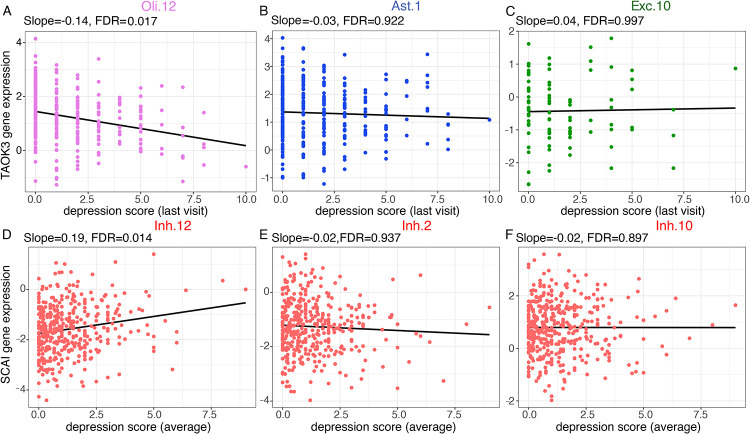
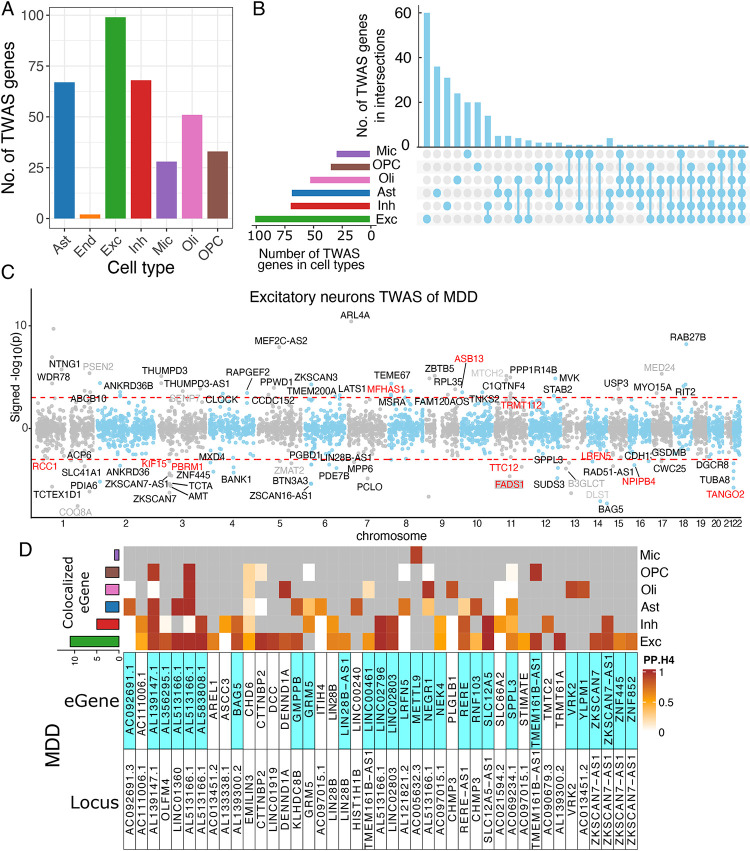
Our single-nucleus TWAS analysis identified 71 causal genes in depression that have a role in specific neocortical cell subtypes; 59 of 71 genes were novel compared to previous studies. Depression TWAS genes showed a cell type specific pattern, with the greatest enrichment being in both excitatory and inhibitory neurons as well as astrocytes. Gene expression in different neuron subtypes have different directions of effect on depression risk. Compared to lower genetically correlated traits (e.g. body mass index) with depression, higher correlated traits (e.g., neuroticism) have more common TWAS genes with depression. In parallel, we performed differential gene expression analysis in relation to depression in 55 cortical cell subtypes, and we found that genes such as , and are associated with depression in specific cell subtypes.
These two sets of analyses illustrate the utility of large snucRNAseq data to uncover both genes whose expression is altered in specific cell subtypes in the context of depression and to enhance the interpretation of well-powered GWAS so that we can prioritize specific susceptibility genes for further analysis and therapeutic development.
抑郁症是一种常见的精神疾病和全球公共卫生问题。然而,我们对抑郁症生物学基础的有限理解阻碍了新型治疗方法和干预措施的发展。
为了确定用于治疗开发的新候选基因,我们研究了来自背外侧前额叶皮质(N = 424)的单核RNA测序(snucRNAseq)数据与生前抑郁症状的关系。为了补充这些直接分析,我们还使用了抑郁症的全基因组关联研究(GWAS)结果(N = 500,199)以及用于推断7种细胞类型和55种细胞亚型中22,159个基因表达的遗传工具,以进行抑郁症的全转录组关联研究(TWAS),随后进行孟德尔随机化(MR)。
我们的单核TWAS分析确定了71个在抑郁症中起作用的因果基因,这些基因在特定的新皮质细胞亚型中发挥作用;与先前的研究相比,71个基因中有59个是新发现的。抑郁症TWAS基因显示出细胞类型特异性模式,在兴奋性和抑制性神经元以及星形胶质细胞中富集程度最高。不同神经元亚型中的基因表达对抑郁风险有不同的影响方向。与与抑郁症遗传相关性较低的性状(如体重指数)相比,遗传相关性较高的性状(如神经质)与抑郁症有更多共同的TWAS基因。同时,我们对55种皮质细胞亚型进行了与抑郁症相关的差异基因表达分析,发现诸如 、 和 等基因在特定细胞亚型中与抑郁症相关。
这两组分析说明了大型snucRNAseq数据在揭示抑郁症背景下特定细胞亚型中表达改变的基因以及增强对强大GWAS的解释方面的效用,以便我们可以确定特定的易感基因进行进一步分析和治疗开发。