School of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, United Kingdom.
Astex Pharmaceuticals, 436 Cambridge Science Park, Milton Road, Cambridge CB4 0QA, United Kingdom.
J Chem Inf Model. 2023 May 8;63(9):2810-2827. doi: 10.1021/acs.jcim.2c01510. Epub 2023 Apr 18.
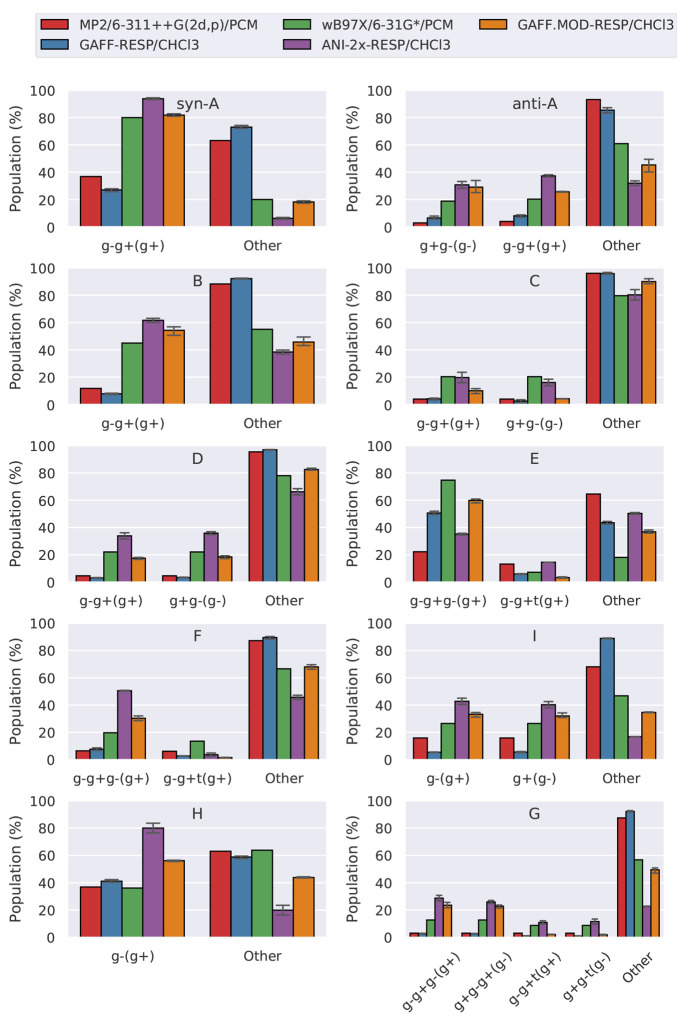
We present a comparative study that evaluates the performance of a machine learning potential (ANI-2x), a conventional force field (GAFF), and an optimally tuned GAFF-like force field in the modeling of a set of 10 γ-fluorohydrins that exhibit a complex interplay between intra- and intermolecular interactions in determining conformer stability. To benchmark the performance of each molecular model, we evaluated their energetic, geometric, and sampling accuracies relative to quantum-mechanical data. This benchmark involved conformational analysis both in the gas phase and chloroform solution. We also assessed the performance of the aforementioned molecular models in estimating nuclear spin-spin coupling constants by comparing their predictions to experimental data available in chloroform. The results and discussion presented in this study demonstrate that ANI-2x tends to predict stronger-than-expected hydrogen bonding and overstabilize global minima and shows problems related to inadequate description of dispersion interactions. Furthermore, while ANI-2x is a viable model for modeling in the gas phase, conventional force fields still play an important role, especially for condensed-phase simulations. Overall, this study highlights the strengths and weaknesses of each model, providing guidelines for the use and future development of force fields and machine learning potentials.
我们进行了一项比较研究,评估了机器学习势(ANI-2x)、传统力场(GAFF)和最佳调谐的 GAFF 类似力场在建模一组 10 种γ-氟醇中的性能,这些分子表现出分子内和分子间相互作用之间的复杂相互作用,从而确定构象稳定性。为了基准化每个分子模型的性能,我们评估了它们相对于量子力学数据的能量、几何和采样准确性。这个基准包括在气相和氯仿溶液中的构象分析。我们还通过将预测值与氯仿中可用的实验数据进行比较,评估了上述分子模型在估计核自旋-自旋耦合常数方面的性能。本研究中的结果和讨论表明,ANI-2x 倾向于预测比预期更强的氢键,并过度稳定全局最小值,并显示出与离域相互作用描述不足相关的问题。此外,虽然 ANI-2x 是气相建模的可行模型,但传统力场仍然发挥着重要作用,特别是对于凝聚相模拟。总体而言,本研究强调了每个模型的优缺点,为力场和机器学习势的使用和未来发展提供了指导。