Giese Timothy J, Huang Ming, Chen Haoyuan, York Darrin M
Center for Integrative Proteomics Research, BioMaPS Institute, and Department of Chemistry and Chemical Biology, Rutgers University , Piscataway, New Jersey 08854-8087 United States.
Acc Chem Res. 2014 Sep 16;47(9):2812-20. doi: 10.1021/ar500103g. Epub 2014 Jun 17.
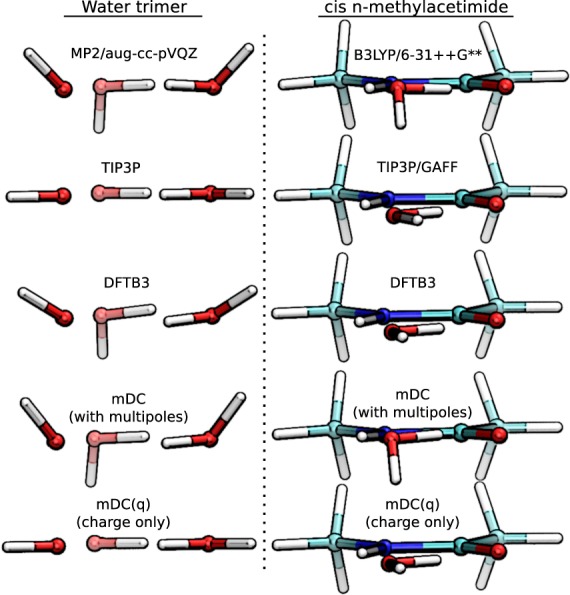
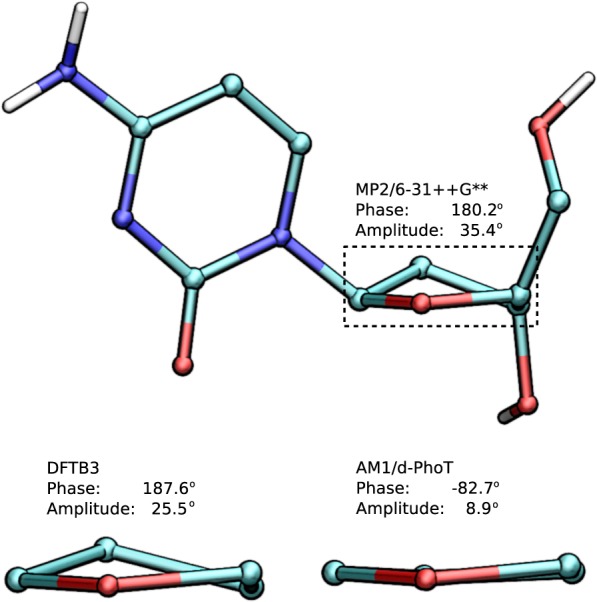
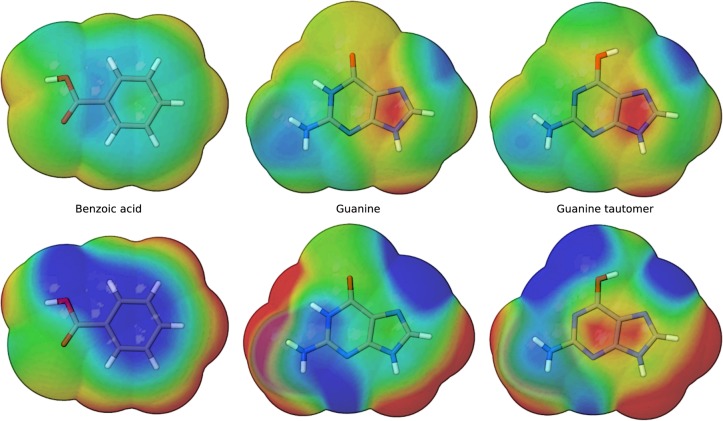
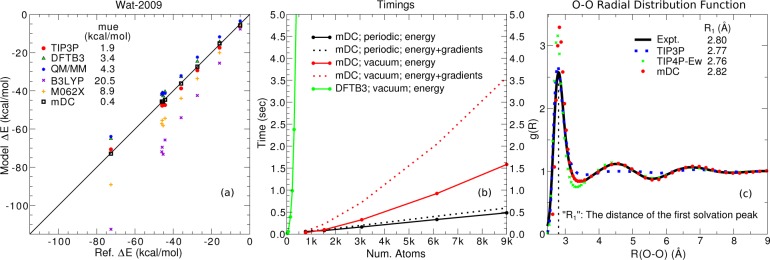
Conspectus There is need in the molecular simulation community to develop new quantum mechanical (QM) methods that can be routinely applied to the simulation of large molecular systems in complex, heterogeneous condensed phase environments. Although conventional methods, such as the hybrid quantum mechanical/molecular mechanical (QM/MM) method, are adequate for many problems, there remain other applications that demand a fully quantum mechanical approach. QM methods are generally required in applications that involve changes in electronic structure, such as when chemical bond formation or cleavage occurs, when molecules respond to one another through polarization or charge transfer, or when matter interacts with electromagnetic fields. A full QM treatment, rather than QM/MM, is necessary when these features present themselves over a wide spatial range that, in some cases, may span the entire system. Specific examples include the study of catalytic events that involve delocalized changes in chemical bonds, charge transfer, or extensive polarization of the macromolecular environment; drug discovery applications, where the wide range of nonstandard residues and protonation states are challenging to model with purely empirical MM force fields; and the interpretation of spectroscopic observables. Unfortunately, the enormous computational cost of conventional QM methods limit their practical application to small systems. Linear-scaling electronic structure methods (LSQMs) make possible the calculation of large systems but are still too computationally intensive to be applied with the degree of configurational sampling often required to make meaningful comparison with experiment. In this work, we present advances in the development of a quantum mechanical force field (QMFF) suitable for application to biological macromolecules and condensed phase simulations. QMFFs leverage the benefits provided by the LSQM and QM/MM approaches to produce a fully QM method that is able to simultaneously achieve very high accuracy and efficiency. The efficiency of the QMFF is made possible by partitioning the system into fragments and self-consistently solving for the fragment-localized molecular orbitals in the presence of the other fragment's electron densities. Unlike a LSQM, the QMFF introduces empirical parameters that are tuned to obtain very accurate intermolecular forces. The speed and accuracy of our QMFF is demonstrated through a series of examples ranging from small molecule clusters to condensed phase simulation, and applications to drug docking and protein-protein interactions. In these examples, comparisons are made to conventional molecular mechanical models, semiempirical methods, ab initio Hamiltonians, and a hybrid QM/MM method. The comparisons demonstrate the superior accuracy of our QMFF relative to the other models; nonetheless, we stress that the overarching role of QMFFs is not to supplant these established computational methods for problems where their use is appropriate. The role of QMFFs within the toolbox of multiscale modeling methods is to extend the range of applications to include problems that demand a fully quantum mechanical treatment of a large system with extensive configurational sampling.

综述 在分子模拟领域,需要开发新的量子力学(QM)方法,以便能够常规应用于复杂、非均匀凝聚相环境中大分子系统的模拟。尽管传统方法,如混合量子力学/分子力学(QM/MM)方法,适用于许多问题,但仍有其他应用需要完全量子力学方法。在涉及电子结构变化的应用中,通常需要QM方法,例如化学键形成或断裂时、分子通过极化或电荷转移相互作用时,或物质与电磁场相互作用时。当这些特征在较大空间范围内出现,在某些情况下可能跨越整个系统时,需要进行完整的QM处理,而不是QM/MM处理。具体例子包括涉及化学键离域变化、电荷转移或大分子环境广泛极化的催化事件研究;药物发现应用,其中广泛的非标准残基和质子化状态用纯经验性的MM力场建模具有挑战性;以及光谱可观测量的解释。不幸的是,传统QM方法巨大的计算成本限制了它们在小系统中的实际应用。线性标度电子结构方法(LSQMs)使大系统的计算成为可能,但计算强度仍然太大,无法在与实验进行有意义比较所需的构型采样程度上应用。在这项工作中,我们展示了适用于生物大分子和凝聚相模拟的量子力学力场(QMFF)开发方面的进展。QMFF利用LSQM和QM/MM方法的优势,产生一种完全QM方法,能够同时实现非常高的精度和效率。QMFF的效率通过将系统划分为片段并在存在其他片段电子密度的情况下自洽求解片段局部分子轨道来实现。与LSQM不同,QMFF引入了经验参数,这些参数经过调整以获得非常精确的分子间力。我们的QMFF的速度和准确性通过从小分子簇到凝聚相模拟的一系列例子以及在药物对接和蛋白质-蛋白质相互作用中的应用得到证明。在这些例子中,与传统分子力学模型、半经验方法、从头算哈密顿量和混合QM/MM方法进行了比较。这些比较证明了我们的QMFF相对于其他模型具有更高的准确性;尽管如此,我们强调QMFF的总体作用不是在其适用的问题上取代这些已确立的计算方法。QMFF在多尺度建模方法工具箱中的作用是扩展应用范围,以包括需要对具有广泛构型采样的大系统进行完全量子力学处理的问题。